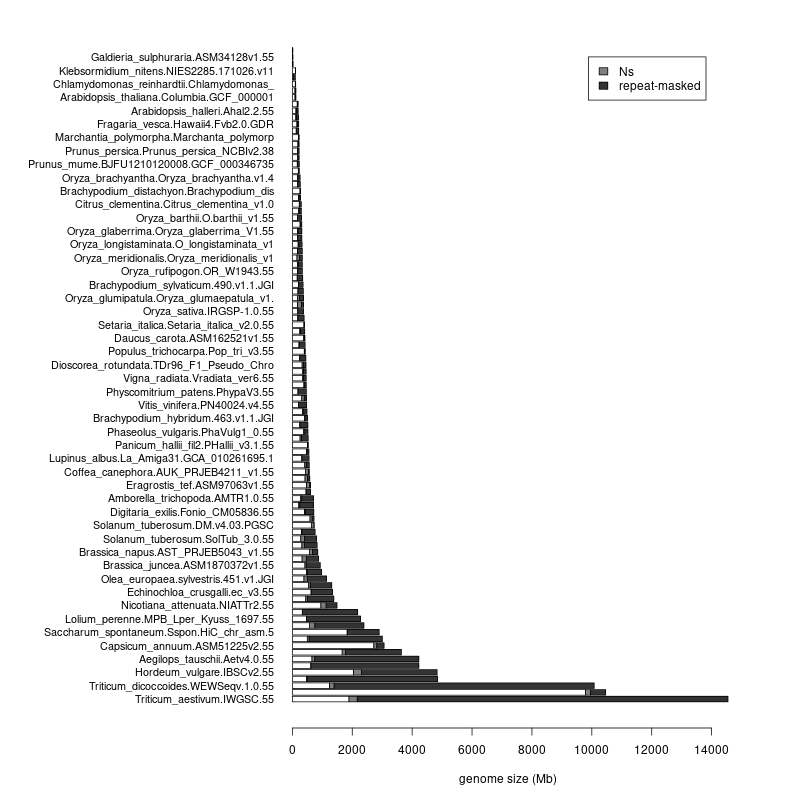
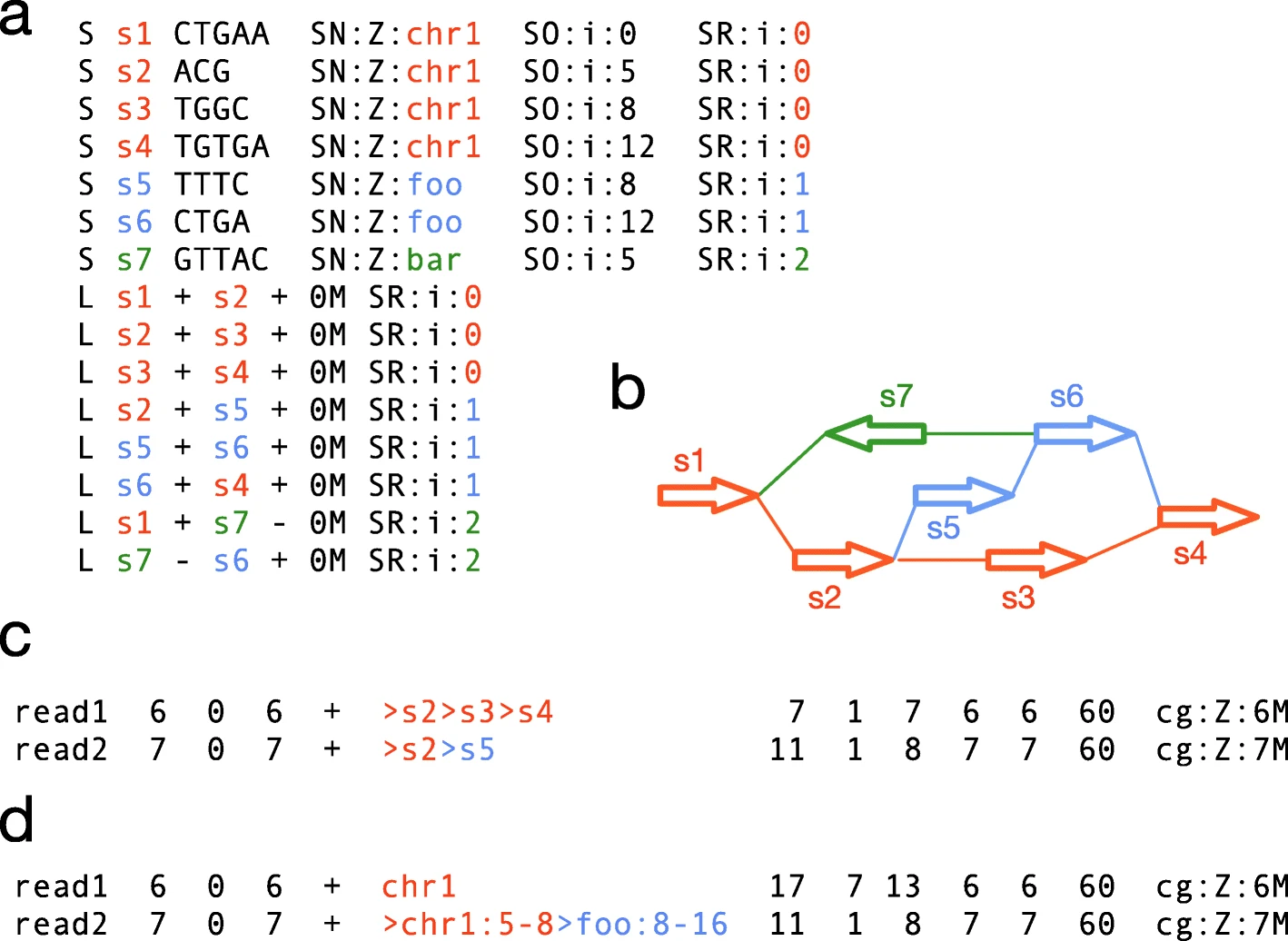
Over the next few days I will be sharing my notes on some of the PAG talks I attend.

Saturday 14012023
This was my first day, you can tell how jet lagged I was as I had to pass on the last session.
Mohamed Zouine, Institut Nationale Polytechinque de Toulouse: when reporting on the quality of a genome assembly it might be useful to use the Assembly Standards of the Earth BioGenome Project, which can be found at https://www.earthbiogenome.org/assembly-standards
Adam William Schoen, University of Maryland: in his group they use MutMap (https://pubmed.ncbi.nlm.nih.gov/22267009/) to find candidate genes for mutant phenotypes. That requires sequencing a single sample of pooled mutant DNA and computing a SNP index, candidate regions have index values close to 1. For instance, the mutant Tin3 has one tiller only in Tmonoc but longer spikes and the candidate gene has 1 SNP in the first position of intron 1, which is then retained.
Matthias Heuberger, University of Zurich: they have a list of genes that seems functional and are expressed in Triticum monoccccum centromeres.
Maria Alejandra Alvarez, University of California, Davis & HHM: she presents data on ELF3 mutants to show that photoperiodic response in wheat does not depend on Ppd1: https://www.biorxiv.org/content/10.1101/2022.10.11.511815v1
Martin Mascher, IPK: Genus-wide pan-genomics. Hordeum bulbosum is ~Myr of evolution away from H. vulgare and can be crossed. It is perennial ourcrossing, the closest wild relative. They are using Hi-C data to assembly separate haplotypes. They typically use Hi-C to check the quality of assemblies. They are now trying pollen sequencing to separate parental genomes of a heterozygous individual. There are larger non-recombining pericentromeric regions in bulbosum than in vulgare. They used a modified, yet unpublished TRITEX protocol which has been used for other species as well. Worth saying these results are actually from a PhD student that could not attend the meeting due to his visa taking a long time, unbelievable this could happen. [HiFi sequencing of barley about 10K per genotype, plus 5K for Hi-C].
Einar Baldvin Haraldsson, Heinrich Heine Universität Düsseldorf. He’s comparing annual barley to perennial Hordeum erectifolium, original from Argentina, which rolls its leaves to keep them erect under drought. IsoSeq with 22 different samples/tissues -> 38K loci, 27K protein coding. Collaborates with MMascher on Pan-Hordeum project (24 species). According to his crosses, perenniality seems to be a dominant trait over annuality.
Eduard Akhunov, Kansas State Univ. He talks about multiplex CRIPR-editing strategies to engineer gene regulation in wheat. Precision is key for multigene families to target the desired copy. He is currently working on deleting regions within promoters of Q gene (an AP TF) and also on silencing clusters of gliadins. He mentions that they needed to resequence the target promoter in the target cultivar, as the CS reference was to divergent to guide their CRISPR constructs. They do both multiplex amplicon and WGS to validate their editing experiments, they have seen to off-targets yet. For the gliadins they used the long-read assembly of cv. Fielder to design gRNA. They confirmed deletions by WGS and also scored protein quality of the edited wheats, with negligible impact of bread making. He says that crosses and field trials are the only way to evaluate the impact of edits in the long term.
William Marande, INRAE – CNRGV: presents the 4Gbp (2Gbp haploid) Vanilla planifolia genome project. Vanilla tissues undergo strict partial endoreplication, which means that C content of cells varies, mixing 2C nuclei with fractions of cells with higher (4,8,16) C contents. So they had to optimize and choose the best starting tissue, which turns out to be auxiliary buds, rich in 2C. When they do K-mer analysis they find an unusual pattern with 3 peaks, as a results of non- and endoreplicated cell populations [as opposed to 2 peaks expected for a heterozygous diploid]. See https://pubmed.ncbi.nlm.nih.gov/35617961 and https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5546068.
Sandra Smit, Bioinformatics Group, Wageningen University and Research. Talks about pangenomes (lossless compressed, integrated, interrogable, scalable, a moving target), which she models with the software PanTools, which is now in version 4.1 and more scalable (hundreds of genomes). It can take for instance all Solanaceae. The latest paper is from 2022 can be found at https://academic.oup.com/bioinformatics/article/38/18/4403/6647839. PanTools is now ploidy-aware and can work at the sequence level as well. They have produced a nice pangenome browser called PanVA: https://www.techrxiv.org/articles/preprint/PanVA_Variant_Analysis_within_Pangenomes/21572433
Wanfang Fu, Department of Plant and Environmental Sciences, Clemson University. She teaches us about extra-chr circular DNAs in plant genomes (eccDNA). They have been proposed to transmit glyphosate resistance in Amaranthus and behave like independent replicons: https://pubmed.ncbi.nlm.nih.gov/36103503, https://academic.oup.com/plcell/article/32/7/2132/6115633. In her work they use the CIDER-Seq protocol to identify them (https://www.nature.com/articles/s41596-020-0301-0), and found a variable number of eccDNAs (in the thousands) in different blackgrass (Alopecurus myosuroides, a weed of wheat) accessions.
Justin N Vaughn, USDA-ARS/University of Georgia, explains how to low-genotype complex loci with variation/pangenome graphs. Mentions phantom SNPs. They are using https://github.com/USDA-ARS-GBRU/PanPipes to build low-cost graphs with Skim-seq (https://www.nature.com/articles/s41598-022-19858-2). Questions whether we need cyclic graphs to capture recombination event? These ideas were applied to melon fungal resistance in https://pubmed.ncbi.nlm.nih.gov/36550124
Isabelle M. Henry, UC Davis. Clonally propagated species are challenging for genomics, such as spearmint (Menta spicata, allotetraploid). She shows nice plots showing coverage vs chr position to highlight regions were there are several haplotypes (higher cover).