Hola,
hace unos meses contruí un pangenoma a partir de 15 genomas de arroz obtenidos de Ensembl Plants. Para ello he probado el software minigraph, descrito en https://doi.org/10.1186/s13059-020-02168-z , que es una de las herramientas disponibles para construir un grafo genómico, en este caso por medio de inserciones y deleciones sobre el genoma de referencia (naranja en la figura).
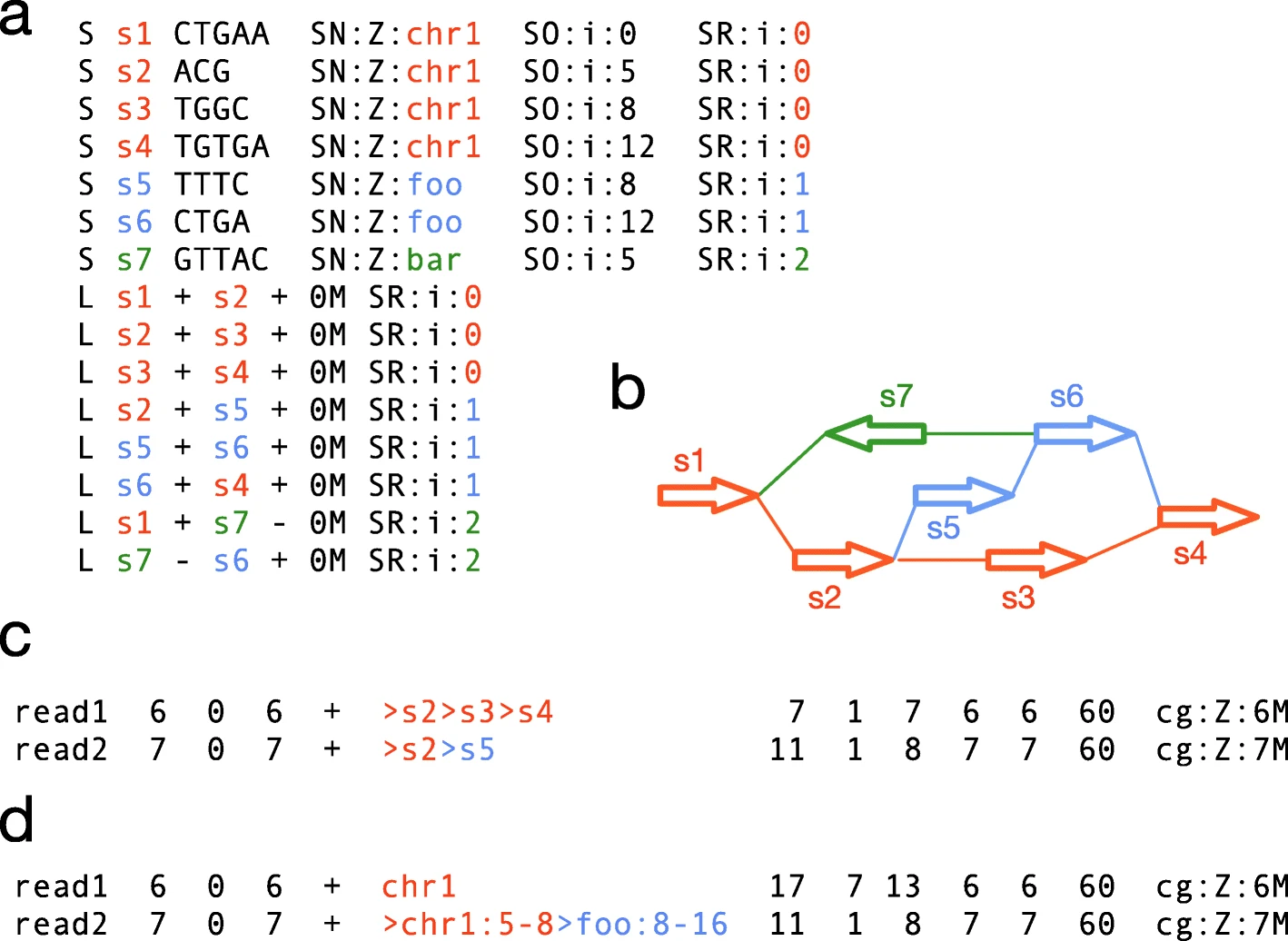 |
| Figura. Mapeo de lecturas sobre un grafo pangenómico, figura tomada de https://doi.org/10.1186/s13059-020-02168-z |
Hoy quería resumir aquí cómo se hace por si le ayuda a alguien.
El primer paso es construir el grafo a partir de varios genomas individuales, de arroz en este caso. Para ello deberás partir de ficheros FASTA donde cada cromosoma tenga un nombre único. Eso se puede lograr por ejemplo agregando al nombre de cromosoma original el identificador o accession de cada genoma:
# 1) prepare genome FASTA files, making sure chr names are unique
mkdir fasta
while read core; do
echo $core;
perl -lne 'BEGIN{ if($ARGV[0] =~ /sativa_([^_]+)/){ $acc=$1 }} if(/^>(\S+)/){ print ">$1_$acc" } else {print}' ${acc}.fna > ${acc}.uniq.fna
done < ../liftover/list_cores.txt
# build the graph
bsub -M 40G -n 10 -cwd soft/minigraph/minigraph -xggs -t 10 oryza_sativa_core_48_101_7.fna fasta/*.fna -o oryza_sativa.gfa
Este proceso genera la siguiente salida:
[M::main::0.702*0.84] loaded the graph from "oryza_sativa_core_48_101_7.fna" [M::mg_index::9.913*1.50] indexed the graph [M::mg_opt_update::10.576*1.47] occ_weight=20, occ_max1=178; 95 percentile: 2 [M::ggen_map::11.491*1.42] loaded file "fasta/oryza_sativa_Azucena_core_48_101_1.fna" [M::ggen_map::168.948*6.05] mapped 37 sequence(s) to the graph [M::mg_ggsimple::170.379*6.01] inserted 15028 events, including 39 inversions [M::mg_index::180.913*5.75] indexed the graph ... [M::main] Real time: 13152.427 sec; CPU: 82007.142 sec; Peak RSS: 47.687 GBAhora podemos probar a mapear secuencias de cDNA sobre el grafo:
# read https://twitter.com/zhigui_bao/status/1417028758725222400 https://github.com/lh3/minigraph/issues/37 # Note in rice -N 0 /-N 100 made no difference! soft/minigraph/minigraph -t 4 -j 0.02 oryza_sativa.gfa -N 100 \ oryza_nivara.cdna.fna | sort -k1,1 -k10,10nr > Onivara.cdna.graph.sort.gaf soft/minigraph/minigraph -t 4 -j 0.02 oryza_sativa.gfa -N 100 \ oryza_sativa.cdna.fna | sort -k1,1 -k10,10nr > Osativa.cdna.graph.sort.gaf soft/minigraph/minigraph -t 4 -j 0.02 oryza_sativa.gfa -N 100 \ oryza_indica.cdna.fna | sort -k1,1 -k10,10nr > Oindica.cdna.graph.sort.gafHasta pronto, Bruno
