Seguro que mapeáis lecturas cortas tipo Illumina como parte de vuestra labor. Si es el caso, entonces conoceréis las herramientas ya clásicas bwa-mem y minimap2, de las que hemos hablado antes (1, 2), desarrolladas por el prolífico Heng Li.
BWA mem se publicó en 2013 destacando su rendimiento al alinear secuencias en torno a los 100b, como las lecturas cortas. Para ser más preciso, nunca se publicó en una revista al ser rechazado, y a día de hoy su preimpresión supera ya las 14K citas, lo que demuestra su utilidad. En cambio, minimap2 alinea con precisión secuencias mucho más largas, como las lecturas HiFi de Pacbio u ONT, o incluso cromosomas enteros. Se publicó en 2018. Como se puede ver en el panel derecho de la Figura 1, ambos algoritmos comparten ideas y componentes y han servido para inspirar otros como bwa-mem2.
 |
| Figura 1. minibwa comparado con otros mapeadores. Fuente: https://x.com/lh3lh3/status/2066917108932329924 |
El mes pasado el autor liberó minibwa, que en sus propias palabras reemplaza a bwa-mem y soporta de manera nativa datos de tipo BS-seq. Podéis ver los detalles de su validación en arxiv, o en éste otro blog, pero en resumen es un algoritmo nuevo que supero limitaciones de diseño de bwa-mem tomando prestadas ideas de minimap2, que además es varias veces más rápido que el primero produciendo resultados muy similares con lecturas cortas. Además, como se ve en la Figura 1 consume mucha menos RAM. En la Figura 2 se muestra que es también mucho más rápido que Bowtie2, otro mapeador muy popular.
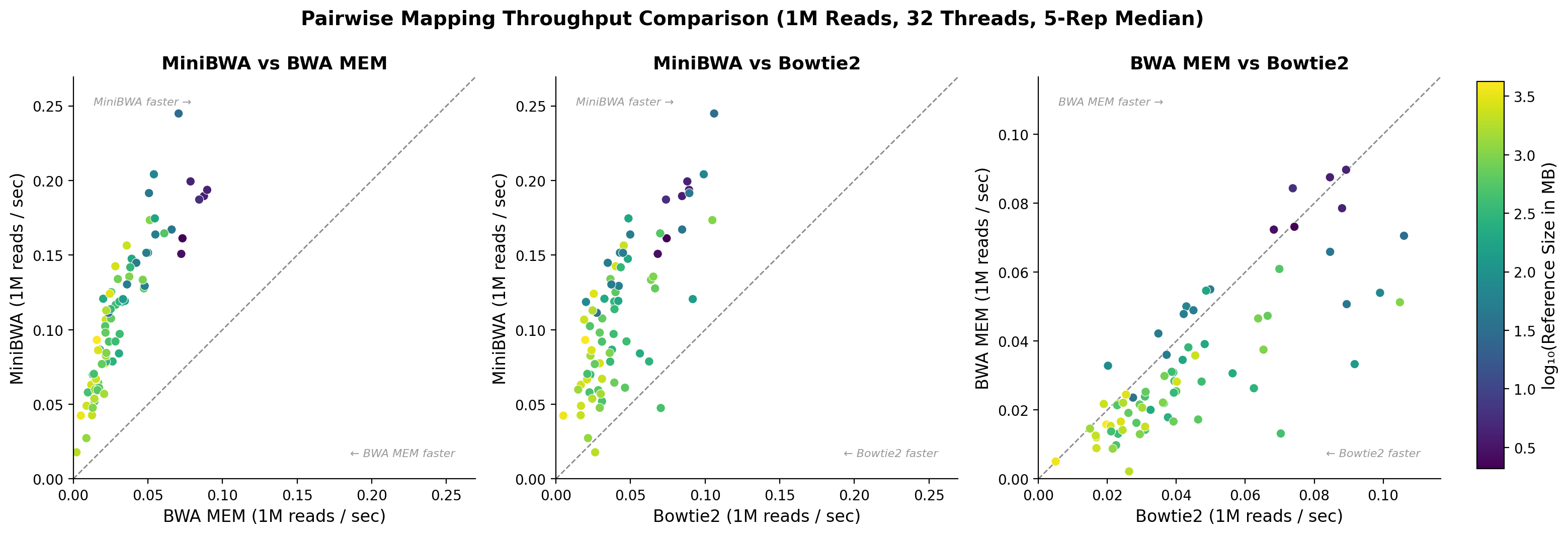 |
| Figura 2. Rendimiento de mapeo de minibwa, bwa-mem y Bowtie2, donce cada punto es un fichero de lecturas cortas de una especie diferente. Fuente: https://andrewcarroll.github.io/2026/06/30/the-best-of-both-worlds-assessing-minibwa.html |
En mis propias pruebas con cebada (4GB) con las versiones bwa-mem 0.7.16a y minibwa 0.6-r416 he observado además:
- El nuevo índice tarda menos en calcularse (minibwa index), supongo que en parte porque se puede paralelizar con varios hilos. Real time: 1633.288 sec; CPU: 2090.351 sec; Peak RSS: 74.990 GB
- El nuevo índice son solamente dos ficheros (.lb2 y .mbw) que en total pesan más (8.9GB) que el índice antiguo (7G).
- Si usas parámetros por defecto puedes cambiar fácilmente un programa por el otro en tus scripts.
El código está disponible en https://github.com/lh3/minibwa,
hasta pronto, Bruno







