Si usas el terminal de linux sabrás de la utilidad de grep para encontrar de manera eficiente cadenas de caracteres o expresiones regulares en ficheros. Si no, tienes ejemplos por ejemplo en nuestro material. En mi caso, aunque usuario habitual, me he tropezado en diferentes ocasiones con el siguiente problema: dame todas las líneas del ficheroA no encontradas en ficheroB. Encontré la mejor solución aquí.
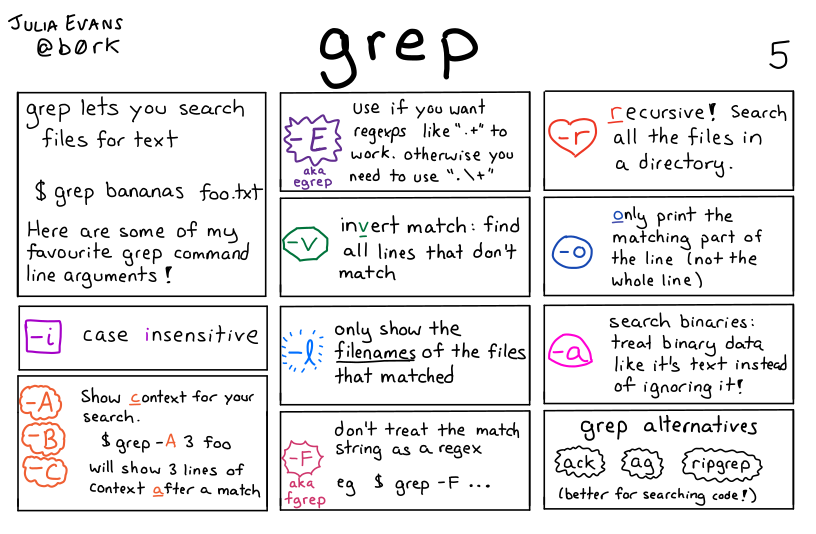 |
| Opciones frecuentes de grep, https://wizardzines.com/comics/grep |
Explico el problema con un ejemplo común en bioinformática. Imagina un fichero con secuencias (ficheroA.fasta) y otro con resultados de análisis de esas secuencias (ficheroB.tsv). Ahora quieres averiguar qué secuencias de A no están presentes en B, por ejemplo para repetir el análisis o arreglar errores en el código.
El ficheroA contiene las siguientes líneas:
>100007_TR35452-c0_g1_i1 CAATTTACGCCTATCGTTATCCATTTCTA... >10000_TR33868-c0_g1_i1 GGGGGACCTACTCAAATCCCCATCTCCC... >10001_TR436-c0_g1_i1 GTTTCCAACCGGATGTTGAAACAGACAA...
El ficheroB en cambio puede ser un formato TSV, por ejemplo:
#metadatos, nombres de columnas, etc 100007_TR35452-c0_g1_i1 chr5H 540009332 540009636 1000_TR868-c0_g2 chr4H 340992292 340995709 ...
Resuelvo el problema en dos comandos en el terminal:
1) extraigo de A únicamente los nombres de las secuencias:
$ perl -lne 'if(/>(\S+)/){print $1}' ficheroA.fasta > ficheroA.nombres
2.i) busco las secuencias reportadas en B para luego 2.ii) buscar las secuencias de A que no están en B:
$ grep -Fo -f ficheroA.nombres ficheroB.tsv | grep -vFf - ficheroA.nombres
Un saludo
