Hola, hoy voy a escribir sobre el software AGC (Assembled Genomes Compressor), escrito en C++, que sirve para comprimir ensamblajes genómicos en múltiples ficheros FASTA (un pangenoma) y luego extraer de manera eficiente secuencias de nucleótidos arbitrarias o contigs completos. El artículo que lo describe está en https://doi.org/10.1093/bioinformatics/btad097 y el código en https://github.com/refresh-bio/agc.
AGC comprime los ensamblajes/ficheros FASTA en varias etapas usando por defecto k=31:
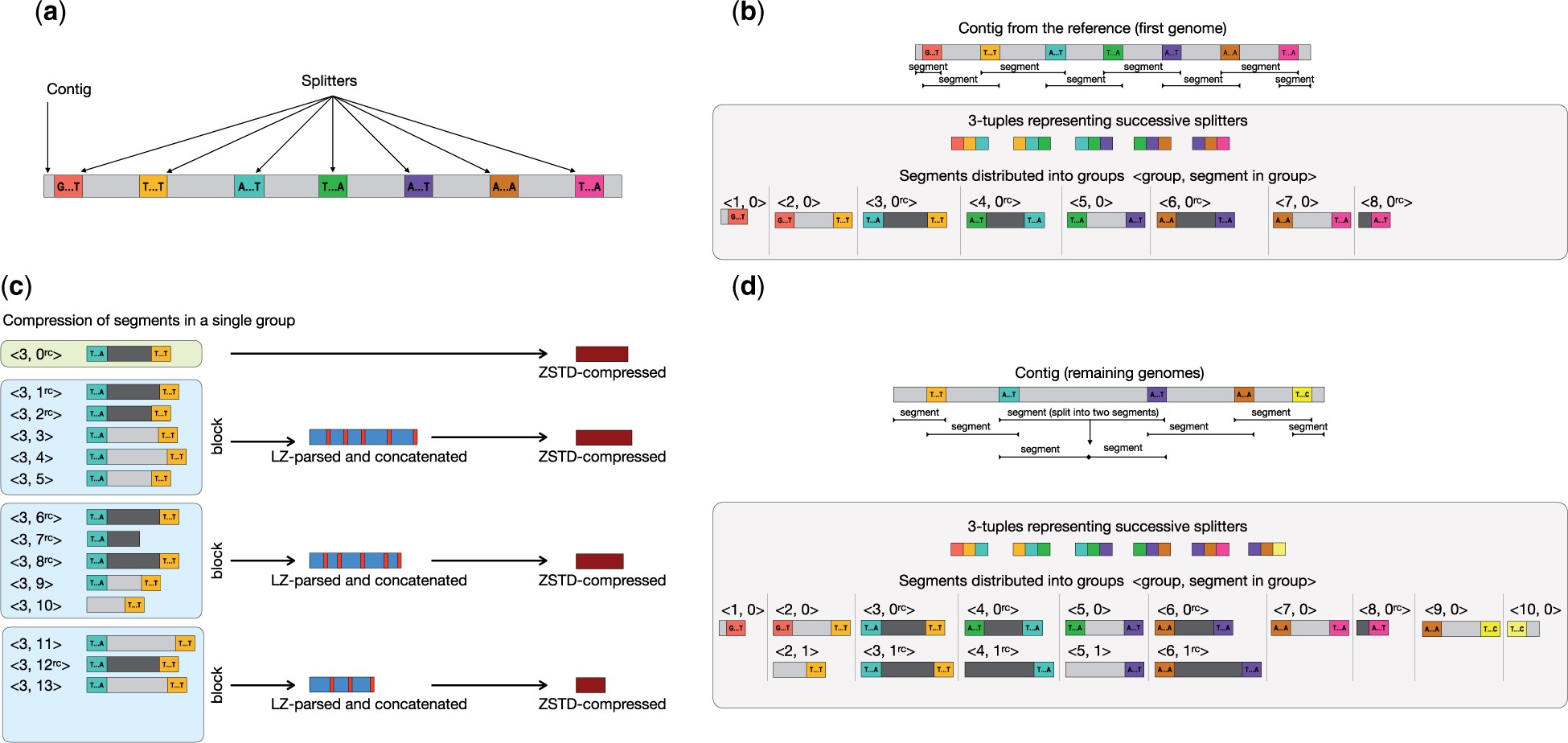 |
Principales etapas de la compresión: (a) selección de k-meros distribuidos de manera uniforme en genoma de referencia cada 60Kb, splitters; (b) compresión del genoma de referencia tras partirlo en grupos y segmentos; (c y d) compresión del resto de genomas. Los segmentos flanqueados por los mismos splitters se comprimen juntos; los de referencia con zstd y los demás con LZSS para marcar las diferencias respecto al de referencia correspondiente para comprimir solamente esas partes con zstd. Figura tomada de https://doi.org/10.1093/bioinformatics/btad097 |
En nuestro caso, probamos AGC con 20 genomas de cebada del pangenoma V2, cada uno de unos 4GB, obteniendo una estructura de datos comprimida de 3.4GB con el siguiente comando:
$ agc create <ref.fa> <genoma2.fa> ... <genoma20.fa> assemblies.agc
# nota: se pueden añadir nuevos genomas más tarde con agc append
Comprobamos el contenido del archivo comprimido:
$ agc listset assemblies.agc
Akashinriki
B1K-04-12
Barke
Chiba
Du_Li_Huang
GoldenPromise
HOR_10350
HOR_13821
HOR_13942
HOR_21599
HOR_3081
HOR_3365
HOR_7552
HOR_8148
HOR_9043
Hockett
Igri
MorexV3
OUN333
Planet
Finalmente, lo más interesante, podemos obtener secuencias de interés por medio de sus coordenadas:
$ agc getctg assemblies.agc chr1H@Barke:1-20
>chr1H sampleName=Barke:1-20
ATGCTATTAGTCACTAATTT
Toda la documentación y más ejemplos están en https://github.com/refresh-bio/agc, que lo disfrutéis,
Bruno












