Hi,
if you use the Plants server of the Regulatory Sequence Analysis Tools (RSAT), you might want to know that it has just been updated. Here's a short summary of the changes:
- The updated URL is https://rsat.eead.csic.es/plants
- It now supports HTTPS connections powered by certbot
- It now uses the source code at https://github.com/rsa-tools/rsat-code (I have updated some documentation along the way)
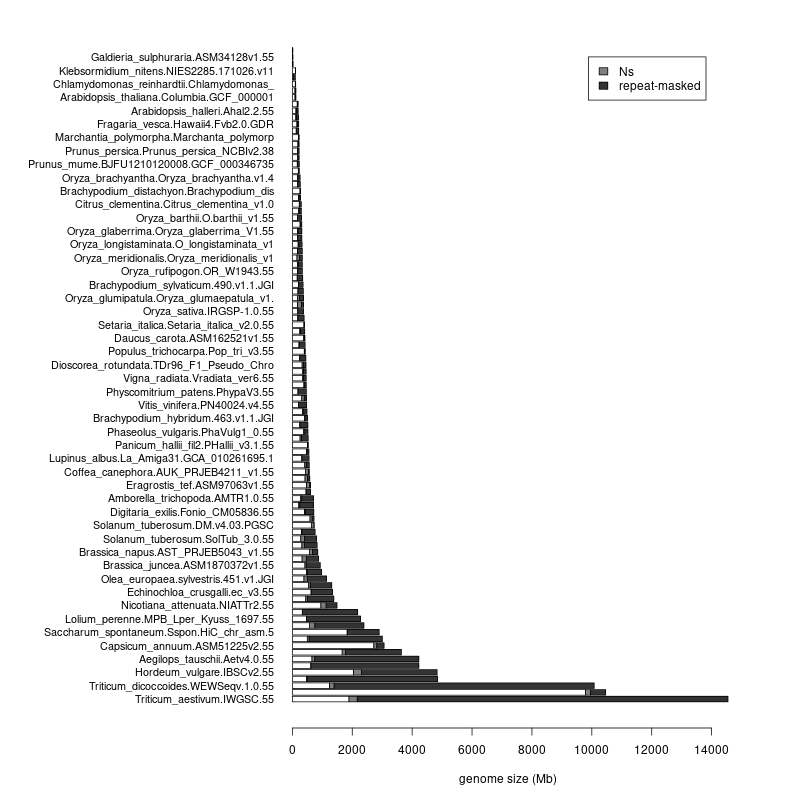
- Nine new species have been imported from Ensembl Plants: Lolium perenne, Brassica juncea, Echinochloa crusgalli, Digitaria exilis, Vigna unguiculata, Brassica rapa ro18, Corylus avellana, Ficus carica, Lactuca sativa
- One species renamed: Physcomitrium patens
- Three updated with a new assembly: Vitis vinifera, Triticum urartum, sunflower
- This leaves the total number of supported assemblies in 100; you can see their stats at https://rsat.eead.csic.es/plants/data/stats
- Most species now correspond to release 55 of Ensembl Plants, but note
that the sequence data is unchanged in many cases. This means that, for
instance, that Hordeum_vulgare.MorexV3_pseudomolecules_assembly.52 becomes Hordeum_vulgare.MorexV3_pseudomolecules_assembly.55, but the sequence is exactly the same.
Have a nice break,
Bruno