Hola,
la semana pasada me encontré con este hilo donde Mo AlQuraishi publicaba que su grupo liberaba por primera vez OpenFold, la primera implementación de AlphaFold2 que podemos ejecutar localmente e incluso re-entrenar con PyTorch, permitiéndonos un control total del proceso. Como dicen los autores en el repositorio, OpenFold es "una reproducción fiel pero entrenable de AlphaFold2". Esto parece resolver la preocupación que expresaba en una entrada anterior sobre cuándo serían estos predictores de código abierto (lee más aquí).
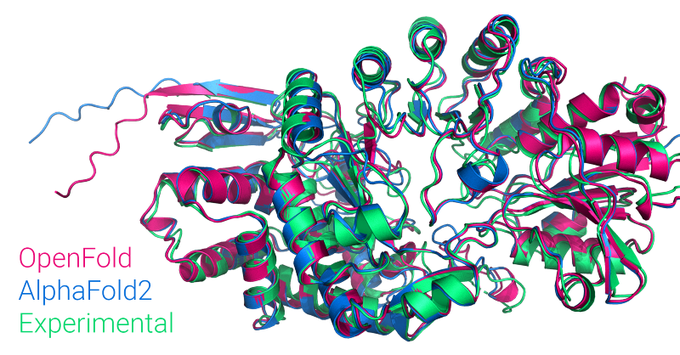
Figura tomada de https://github.com/aqlaboratory/openfold
Como explica Mo, también han puesto a disposición de cualquiera los datos que usaron para entrenar OpenFold, siguiendo exactamente la metodología del artículo original de AlphaFold2. El código está disponible en https://github.com/aqlaboratory/openfold y se puede probar de varias maneras:
- un cuaderno Colab (similar a ColabFold)
- un contenedor Docker
- un script python local con instrucciones de cómo hay que instalar las dependencias y los datos, un proceso largo y farragoso, pero el más adecuado para sacar partido a tu GPU
Hasta pronto,
Bruno


