Hola,
a pesar de que lamentablemente la guerra sigue en Ucrania, hoy continuamos el marcaje que hacemos desde este blog a las enzimas Cas9 y las secuencias CRISPR (ver por ejemplo esta entrada), porque la actualidad nos ha traído novedades. Pero vayamos por partes.
La primera novedad es un trabajo de ingeniería de proteínas (publicado en https://doi.org/10.1038/s41586-022-04470-1) donde los autores avanzan en la comprensión del mecanismo de corte de la enzima Cas9 (Figura 1) y lo aprovechan para hacer una mutagénesis dirigida donde reemplazan algunos aminoácidos para que dejen de estabilizar los nucleótidos 18-20 del ADN diana en caso de no aparear (Figura 2) y sin afectar a la velocidad de la reacción de las secuencias apareadas:
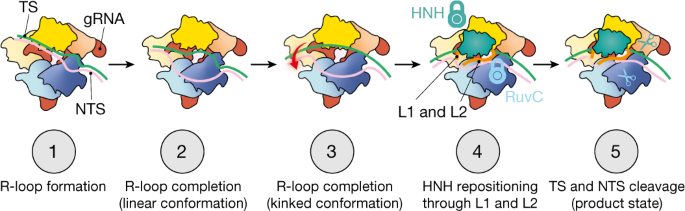
Figura 1. Modelo para la activación de la enzima Cas 9 tomado de https://doi.org/10.1038/s41586-022-04470-1.

Figura 2. Esquema del complejo PAM-distal gRNA–TS con moléculas de agua como círculos rojos. Los aminoácidos que contactan con los nucleótidos C18, A19 y G20 fueron mutados para comprobar su efecto en el corte de la secuencia diana. Las curvas muestran estudios de la dinámica de la reacción enzimática de corte de la Cas9 comparando moléculas guía que aparean (On-target) con moléculas que contienen mismatches (MM, Off-target) en las posiciones 18-20. Adaptada de https://doi.org/10.1038/s41586-022-04470-1.
Qué significa esto? Pues que esta versión de Cas9 (SuperFi-Cas9) tiene mayor fidelidad y es un paso adelante en la dirección de conseguir enzimas que no corten donde no se esperaban cortes.
La segunda novedad es que en la batalla legal por los derechos de explotación de las tecnologías CRISPR parece de momento ha ganado la Universidad de Harvard, como podéis leer por ejemplo en https://www.technologynetworks.com/genomics/news/broad-institute-wins-crispr-patent-case-359160 . Ya veremos que consecuencias tiene esto para el uso de las tecnologías Cas9 en ciencia, pero vemos que si los premios Nobel dejaron fuera a algunos investigadoes clave como Francis Mójica (ver por ejemplo esto), ahora la guerra de las patentes deja fuera a las investigadoras que ganaron el Nobel por este trabajo!
Hasta pronto,
Bruno


