Hace un año Homma, Huang y van der Hoorn publicaron en Nature Comms sus experimentos modelando complejos de proteínas híbridos planta:patógeno con AlphaFold-Multimer (AFM). En concreto, encontraron una manera de encontrar SSPs, proteínas pequeñas secretadas por microorganismos patógenos de plantas que se unen de manera específica a proteínas de la planta diana. En total, su cribado con AFM consideró las combinaciones de 1879 SSPs de bacterias y hongos patógenos del tomate y 6 proteasas endógenas que participan en la defensa frente a la infección:
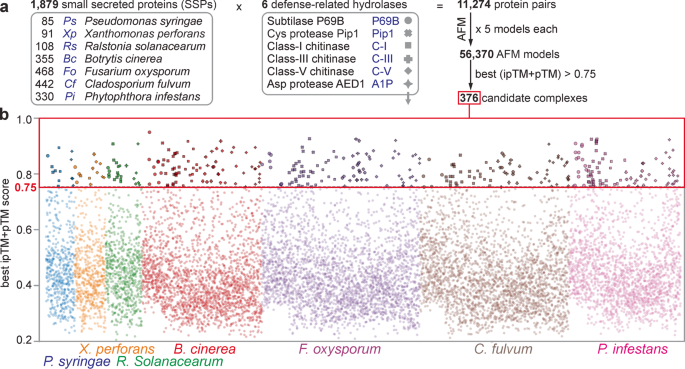 |
| Modelos de parejas de proteínas planta:patógeno modelados con AFM que superan el umbral 0.75, tomada de https://doi.org/10.1038/s41467-023-41721-9. |
De 376 complejos proteína:proteína prometedores, elegidos por sus puntuaciones ipTM+pTM, se centraron en 15 complejos donde SSPs sin anotar bloqueaban los sitios activos de quitinasas y proteasas de tomate. De esos, encontraron confirmación experimental para 4.
Dado el interés que despertaron estos resultados, los mismos autores han publicado ahora un protocolo (https://doi.org/10.1111/tpj.16969) para hacer este tipo de predicciones usando ColabFold en la Web y localmente (leer más en blog).
El protocolo tiene los siguientes pasos:
- Start with ColabFold online
- Use a computing cluster for screens
- Small sequences model faster
- Curate the input sequences
- Remove irrelevant domains
- Include positive controls
- Include negative controls
- Recycle multiple sequence alignments (MSAs)
- Control data storage
- Separate CPU from GPU-intense steps
- Try to get MSA >100
- Evaluate the predicted scores
- Beware of typical AFM errors
- Beware of false negatives
- Beware of false positives
- Explore hits manually
- Categorise hits in classes

Hasta pronto,
Bruno

