Hola,
la pandemia que estamos viviendo, un año después, nos está poniendo a prueba. A pesar de la improvisación de los políticos a escala global, de la desinformación en las redes sociales y los brotes de desconfianza, a pesar de la economía bajo mínimos y del cole en casa, ahora la mayoría tenemos la esperanza de que las vacunas resuelvan el problema.
En este artículo solamente pretendo recordar el largo camino de la investigación básica que nos ha traído hasta el presente. Estas vacunas son ya grandes hitos de la humanidad, a la altura de la llegada a la luna, pero el camino ha sido largo, de al menos 25 años. Por tanto, nada de milagros, son el fruto de mucho trabajo acumulado que fue explotado con mucho éxito por empresas como BioNTech y Moderna. Como pasó con CRISPR para la edicion de genomas, para que alguien llegara a la cima fueron necesarios muchos pasos previos, muchos de los cuales fuera de contexto serían objeto de "y eso para qué sirve". Aquí enumero los más importantes para las vacunas de ARN, extraídos de este hilo (no están los de los otros tipos de vacunas):
1970 T7 ARN polimerasa: nature.com/articles/22822 . Esta enzima permite sintetizar moléculas de ARN a medida, como las de las vacunas.
1978 Liposomas para llevar ARN mensajeros (mRNA): nature.com/articles/27492 . Estos vehículos permiten que el ARN de las vacunas pueda atravesar
la doble capa lipídica de la membrana celular.
1990 Inyecciones de ADN y ARN para expresar genes de manera transitoria en tejidos: science.sciencemag.org/content/247/49 . Este trabajo demostró que es posible expresar genes a medida tras ser inyectados en tejidos, de manera que se traducen como proteínas.
2005 Ribonucleótidos modificados no disparan respuestas inmunes: cell.com/immunity/fullt .
Esto permite que el ARN inyectado no desencadene una reacción inmune por si mismo, lo que se pretende es que la reacción la desencadene la proteína codificada por ese ARNm.
2017 Estabilización de proteínas expuestas de los coronavirus MERS-CoV y SARS-CoV: pnas.org/content/114/35. Esto permite que la proteína modificada del coronoavirus que expresa el ARNm sea más estable y desencadene una reacción inmune más robusta.
Esta sucesión de descubrimientos y la tecnología actual permitieron que el tiempo de desarrollo de las vacunas haya sido el más corto de la historia:
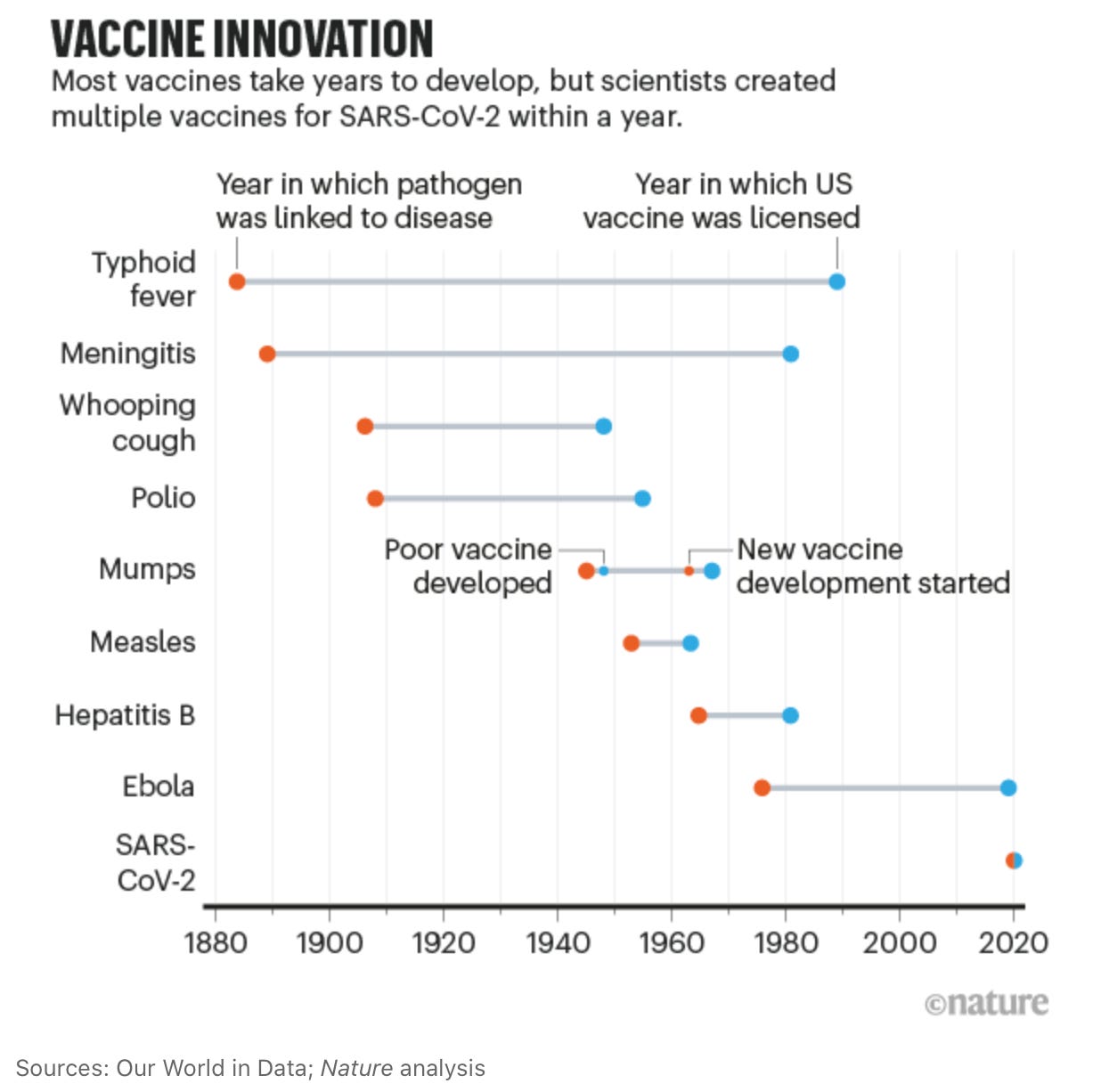
En mi actual institución, el Instituto Europeo de Bioinformática (EBI), también hemos contribuído con el https://www.covid19dataportal.org y el navegador de genomas https://covid-19.ensembl.org
Espero haberos convencido de que la ciencia financiada con fondos públicos y transparente, muchas veces invisible e ingrata, es una parte fundamental e integral del avance de nuestra sociedad, y que de ella beben las empresas que nos venden luego los productos.
Hasta pronto,
Bruno
