[He movido el contenido de esta entrada a la página de ofertas laborales]
23 de enero de 2015
15 de enero de 2015
La PCR más grande del mundo
En la entrada de hoy veremos la utilidad del comando de linux AWK, que fue utilizado en el anterior post para extraer secuencias aleatorias de un archivo FASTA o FASTQ.
AWK es más que un comando, es un poderoso lenguaje usado para manipular y procesar grandes ficheros de texto. Si queréis más información, en internet hay muy buenos tutoriales.
GAWK es la versión GNU de AWK que usan todas las distribuciones linux, se puede ver que 'awk' en Ubuntu redirige a 'gawk':
A lo que íbamos, el título del post es 'La PCR más grande del mundo' porque la idea es mostrar cómo hacer una PCR in silico usando AWK.
Para ello prepararemos 3 ficheros, uno con los primers, otro con las secuencias con las que vamos a simular la PCR (ambos en formato FASTA) y otro con el código AWK.
Ejemplo de fichero 'primers.fa':
Observar que hay que poner el primer directo en sentido 5'->3' y el reverso en sentido 3'->5', tal como los veríamos en la secuencia 5'->3' del archivo 'secuencias.fa' donde los vamos a buscar.
Ejemplo de fichero 'secuencias.fa':
Fichero con el código AWK 'pcr.awk':
Y ejecutaremos el siguiente comando:
O en un sola línea podemos poner todo el código AWK:
El resultado será el siguiente:
Cada columna indica lo siguiente:
ACTUALIZACIÓN:
Probando un código similar escrito en Perl (gracias Bruno por el apunte) descubrimos que Perl es mucho más rápido que AWK y sólo tarda 1 minuto medio!!!
Código en Perl en el fichero 'pcr.pl':
AWK es más que un comando, es un poderoso lenguaje usado para manipular y procesar grandes ficheros de texto. Si queréis más información, en internet hay muy buenos tutoriales.
GAWK es la versión GNU de AWK que usan todas las distribuciones linux, se puede ver que 'awk' en Ubuntu redirige a 'gawk':
$ which awk
/usr/bin/awk
$ ls -l /usr/bin/awk
/usr/bin/awk -> /etc/alternatives/awk
$ ls -l /etc/alternatives/awk
/etc/alternatives/awk -> /usr/bin/gawk
A lo que íbamos, el título del post es 'La PCR más grande del mundo' porque la idea es mostrar cómo hacer una PCR in silico usando AWK.
Para ello prepararemos 3 ficheros, uno con los primers, otro con las secuencias con las que vamos a simular la PCR (ambos en formato FASTA) y otro con el código AWK.
Ejemplo de fichero 'primers.fa':
>Marker_1
GTTGTGTCTTTAGCTCCCCTG(.+)TA[G|T]ATCAGGTGAGCCGAT
>Marker_2
ACTACAACCAGAGCGAGG(.+)AG[A|T]TC[C|G]CCCAAAGGCACA
Observar que hay que poner el primer directo en sentido 5'->3' y el reverso en sentido 3'->5', tal como los veríamos en la secuencia 5'->3' del archivo 'secuencias.fa' donde los vamos a buscar.
Ejemplo de fichero 'secuencias.fa':
>QK4PW:00814:02853
AAGACAGTTGTGTCTTTAGCTCCCCTGAGCTGAACGACATCAGTACATCAGGTCCAACTTCTACAACAAGCTGGAGCTTTTCAGGTTTGACAGCAACCTGGGGAGTTTGTTGGATACACAGAATATGGAGTGAAACAAGCTGAATACAGGAACAACAACCCGTCATATATCGCATCACTGAGAGCTCGAGAGGGCAGACCGCCTGCCTGCACAACATTGGTATTGACTACCAGAACATTCTGACTAGATCAGGTGAGCCGATTCGGTT
>QK4PW:03633:02817
TCACTCGTTGTGTCTTTAGCTCCCCTGAGCTGAAGGACATTCAGTACATCAACTCCTATTATTACAACAAGCTGGAATGGGCCAGGTTTGACAGCAACGTGGGTAGATATGTTGGATACACGAAGTTCGGAGTTTATAATGCAGAACGATGGAACAAAGACCCGTCAGAGATCGCTATGAGGATGGCTCAGAGGGAGACCTACTGCCTGCACAACATTGGTAACTGGTACCCAAACATGCTGACTAGATCAGGTGAGCCGATCGCGTT
>QK4PW:01993:02583
CACAGTGTTGTGTCTTTAGCTCCCCTGAGCTCAAAGACATCCAGTACATCAGGTCCTATTATTACAACAAGCTGGAGTTCATCAGGTTTGACAGCAACGTGGGGGAGTTTGTTGGATACACGGAGCTGGGAGTGAAGAACGCTGAGCGGCTCAACAACGACCCGTCACAGATCGCTGGGTTGAAAGCTCAGAGGGAGGCCTACTGCATGAACCATGTTACTGCTTTCTACCCAAACGCTCTGGATATATCAGGTGAGCCGATCAGCGG
>QK4PW:01336:01442
CTCATGACTACAACCAGAGCGAGGGCGGCTCTCACACCATCCAGGAGATGTATGGCTGTGACGTGGGGTCGGACGGGCGCCTCCTCCGCGGGTACAGTCAGTTCGCCTACGATGGCCGCGATTACATCGCCCTGAACGAGGACCTGACGACATGGACGGCGGCGGACACGGCGGCGCAGATCTCCCAGCGCAAGTTGGAGCAGGCTGGTGCTGCTGAGATATACAAGGCCTACCTGGAGGACGAGTGCGTGCAGTGGCTGCGCAGACACCTGGAGAACGGGAAGGGACGAGCTGCTGCGCACAGTTCGCCCAAAGGCACAATGTTA
>QK4PW:00383:00875
TCGACGACTACAACCAGAGCGAGGGCGGCTCTCACACCATCCAGGAGATGTATGGCTGTGACGTGGGGTCGGACGGGCGCCTCCTCCACGGGTATTATCAGTTCGCCTACGACGGCCGCGATTACATCGCCCCTGAACGAGGACCTGAAGACGTGGACGGCAGCGGACACGGCGGCGCAGATCACCCAGCGCAAGTGGGAGCAGGCTGGTGATGCAGAGAGTCTGAAGGCCTTCCTGGAGGGCGAGTACACGCAGTGGCTGCGCAGATTCCTGGAGATCGGGAAGGACGCGCTGCTGCGCACAGTTCCCCCAAAGGCACACACATA
Fichero con el código AWK 'pcr.awk':
# Primero leemos el archivo FASTA con los primers
# Y guardamos las secuencias de los primers en un array
FNR==NR{
if ($0~/>/){
primer_name=$0;
} else if ($0) {
primers[primer_name]=$0;
}
next;
}
# Después procesamos el archivo FASTA de secuencias
# Buscando los primers en cada secuencia
{
if ($0~/>/) {
query_name=$0;
} else if ($0) {
for (primer_name in primers) {
if (p=match($0, primers[primer_name])) {
print query_name"\t"primer_name"\t"RSTART"\t"RLENGTH"\t"substr($0, RSTART, RLENGTH);
}
}
}
}
Y ejecutaremos el siguiente comando:
$ awk -f pcr.awk primers.fa secuencias.fa
O en un sola línea podemos poner todo el código AWK:
$ awk 'FNR==NR{ if ($0~/>/){primer_name=$0;} else if ($0) {primers[primer_name]=$0;} next;} { if ($0~/>/) {query_name=$0;} else if ($0) { for (primer_name in primers) { if (p=match($0, primers[primer_name])) { print query_name"\t"primer_name"\t"RSTART"\t"RLENGTH"\t"substr($0, RSTART, RLENGTH); } } } }'primers.fa secuencias.fa
El resultado será el siguiente:
>QK4PW:00814:02853 >Marker_1 7 256 GTTGTGTCTTTAGCTCCCCTGAGCTGAACGACATCAGTACATCAGGTCCAACTTCTACAACAAGCTGGAGCTTTTCAGGTTTGACAGCAACCTGGGGAGTTTGTTGGATACACAGAATATGGAGTGAAACAAGCTGAATACAGGAACAACAACCCGTCATATATCGCATCACTGAGAGCTCGAGAGGGCAGACCGCCTGCCTGCACAACATTGGTATTGACTACCAGAACATTCTGACTAGATCAGGTGAGCCGAT
>QK4PW:03633:02817 >Marker_1 7 256 GTTGTGTCTTTAGCTCCCCTGAGCTGAAGGACATTCAGTACATCAACTCCTATTATTACAACAAGCTGGAATGGGCCAGGTTTGACAGCAACGTGGGTAGATATGTTGGATACACGAAGTTCGGAGTTTATAATGCAGAACGATGGAACAAAGACCCGTCAGAGATCGCTATGAGGATGGCTCAGAGGGAGACCTACTGCCTGCACAACATTGGTAACTGGTACCCAAACATGCTGACTAGATCAGGTGAGCCGAT
>QK4PW:01993:02583 >Marker_1 7 256 GTTGTGTCTTTAGCTCCCCTGAGCTCAAAGACATCCAGTACATCAGGTCCTATTATTACAACAAGCTGGAGTTCATCAGGTTTGACAGCAACGTGGGGGAGTTTGTTGGATACACGGAGCTGGGAGTGAAGAACGCTGAGCGGCTCAACAACGACCCGTCACAGATCGCTGGGTTGAAAGCTCAGAGGGAGGCCTACTGCATGAACCATGTTACTGCTTTCTACCCAAACGCTCTGGATATATCAGGTGAGCCGAT
>QK4PW:01336:01442 >Marker_2 7 314 ACTACAACCAGAGCGAGGGCGGCTCTCACACCATCCAGGAGATGTATGGCTGTGACGTGGGGTCGGACGGGCGCCTCCTCCGCGGGTACAGTCAGTTCGCCTACGATGGCCGCGATTACATCGCCCTGAACGAGGACCTGACGACATGGACGGCGGCGGACACGGCGGCGCAGATCTCCCAGCGCAAGTTGGAGCAGGCTGGTGCTGCTGAGATATACAAGGCCTACCTGGAGGACGAGTGCGTGCAGTGGCTGCGCAGACACCTGGAGAACGGGAAGGGACGAGCTGCTGCGCACAGTTCGCCCAAAGGCACA
>QK4PW:00383:00875 >Marker_2 7 314 ACTACAACCAGAGCGAGGGCGGCTCTCACACCATCCAGGAGATGTATGGCTGTGACGTGGGGTCGGACGGGCGCCTCCTCCACGGGTATTATCAGTTCGCCTACGACGGCCGCGATTACATCGCCCCTGAACGAGGACCTGAAGACGTGGACGGCAGCGGACACGGCGGCGCAGATCACCCAGCGCAAGTGGGAGCAGGCTGGTGATGCAGAGAGTCTGAAGGCCTTCCTGGAGGGCGAGTACACGCAGTGGCTGCGCAGATTCCTGGAGATCGGGAAGGACGCGCTGCTGCGCACAGTTCCCCCAAAGGCACA
Cada columna indica lo siguiente:
- Nombre de la secuencia que unen los primers.
- Nombre de la pareja de primers.
- Posición de inicio de la amplificación.
- Longitud de la cadena amplificada.
- La secuencia amplificada.
ACTUALIZACIÓN:
Probando un código similar escrito en Perl (gracias Bruno por el apunte) descubrimos que Perl es mucho más rápido que AWK y sólo tarda 1 minuto medio!!!
perl pcr.pl primers.fa secuencias.faCódigo en Perl en el fichero 'pcr.pl':
open(PRIMERFILE, $ARGV[0]);
while(<PRIMERFILE>){
chomp;
if (/>/){
$primer_name=$_;
} elsif ($_) {
$primers{$primer_name}=$_;
}
}
close PRIMERFILE;
open(SEQSFILE, $ARGV[1]);
while(<SEQSFILE>){
chomp;
if (/>/){
$query_name=$_;
} elsif ($_) {
foreach my $primer_name (keys %primers) {
if (/$primers{$primer_name}/) {
printf("%s\t%s\t%s\t%s\t%s\n",$query_name,$primer_name,$-[0]+1,length($&),$&);
}
}
}
}
close SEQSFILE;
Para finalizar siento decir que esto no es nada nuevo, existen numerosos programas y utilidades web para realizar simulaciones de PCR a partir de un conjunto de primers y un fichero FASTA de secuencias, pero creo que ninguno tan sencillo y rápido como el código de AWK para procesar millones de secuencias:
Artículo homenaje al inventor de la PCR, Kary Mullis.

- UCSC In-Silico PCR: Además de realizar la PCR permite controlar el tamaño de las secuencias amplificadas y el número mínimo de bases exactas en 3' para cada primer. El enlace nos lleva a su versión de servidor web, se puede descargar aquí. Debo decir que su versión en línea de comandos no me ha dado buenos resultados.
- e-PCR: permite simular PCRs y encontrar secuencias de DNA que serían reconocidas de forma específica y no específica por un conjunto de primers. Posee versión web y descargable.
- EHU In-Silico PCR: Permite con su interfaz web seleccionar un genoma, un par de primers y obtener los productos de los mismos.
- FastPCR: un programa comercial para el diseño de primers y su testeo in silico. Quizás sea el programa más completo si estamos interesados en el diseño de PCRs y nuestro laboratorio puede comprar el software.
- Primer-BLAST: sirve para diseñar primers y probarlos contra un fichero de secuencias por medio de BLAST para buscar todos los posibles productos amplificados (normalmente buscaremos subproductos generados por uniones no específicas de los primers).
- Simulate_PCR: un script en Perl que tomo como entrada un listado de primers y un fichero FASTA y devuelve los posibles productos de PCR usando alineamientos de BLAST.
- GASSST (Global Alignment Short Sequence Search Tool): genera alineamientos globales de un conjunto de primers contra un fichero de secuencias. Parseando sus resultados con un script podremos reconstruir productos amplificados por uniones específicas y no-específicas de los primers. La dificultad de tener que procesar los resultados es su principal problema, pero realiza alineamientos globales de secuencias muy cortas (como los primers) que no consigue casi ningún programa.
Artículo homenaje al inventor de la PCR, Kary Mullis.
9 de enero de 2015
Algunos comandos útiles de linux para manejar ficheros FASTQ de NGS
 Leyendo la entrada de Bruno sobre la librería kseq.h me entraron ganas de recopilar en el blog unos valiosos comandos de Linux que nos pueden ayudar a salvar mucho tiempo al trabajar con millones de secuencias de NGS.
Leyendo la entrada de Bruno sobre la librería kseq.h me entraron ganas de recopilar en el blog unos valiosos comandos de Linux que nos pueden ayudar a salvar mucho tiempo al trabajar con millones de secuencias de NGS.Un fichero con extensión '.fq.gz' es un fichero en formato FASTQ comprimido en formato GZIP. Los ficheros FASTQ con datos de los nuevos métodos de secuenciación (NGS) suelen ocupar decenas de Gigabytes de espacio de disco (una cantidad indecente para la mayoría de los mortales) y comprimidos con GZIP se reducen (mucho más que con el clásico ZIP).
A continuación se mostrarán los comandos para manejar ficheros FASTQ comprimidos, pero con unas leves modificaciones podrán también procesar ficheros FASTQ sin comprimir (no recomendado), por ejemplo cambiando 'zcat' por 'cat'. También se pueden modificar para procesar ficheros FASTA comprimidos y sin comprimir.
Para empezar, vamos a comprimir en formato GZIP un archivo FASTQ:
> gzip reads.fq
El fichero resultante se llamará por defecto 'reads.fq.gz'.
Nos puede interesar conocer cuanto espacio ocupa realmente un archivo 'fq.gz' comprimido, para ello usaremos el mismo comando 'gzip':
> gzip --list reads.fq.gz
compressed uncompressed ratio uncompressed_name
18827926034 1431825024 -1215.0% reads.fq
Parece que GZIP en vez de comprimir expande, pero no es verdad, simplemente que la opción '--list' de 'gzip' no funciona correctamente para archivos mayores de 2GB. Así que hay que recurrir a un método más lento y esperar unos minutos:
> zcat reads.fq.gz | wc --bytes
61561367168
Si queremos echar un vistazo al contenido del archivo podemos usar el comando 'less' o 'zless':
> less reads.fq.gz
> zless reads.fq.gz
Y para saber el número de secuencias que contiene simplemente hay que contar el número de líneas y dividir por 4 (le costará unos minutos):
> zcat reads.fq.gz | echo $((`wc -l`/4))
256678360
Contar el número de secuencias en un fichero FASTA:
> grep -c "^>" reads.fa
Podemos contar cuántas veces aparece una determinada secuencia, por ej. ATGATGATG:
> zgrep -c 'ATGATGATG' reads.fq.gz
398065
> zcat reads.fq.gz | awk '/ATGATGATG/ {nlines = nlines + 1} END {print nlines}'
398065
O extraer ejemplos de dicha secuencia:
> zcat reads.fq.gz | head -n 20000 | grep --no-group-separator -B1 -A2 ATGATGATG
en ficheros FASTA:
> zcat reads.fa.gz | head -n 10000 | grep --no-group-separator -B1 ATGATGATG
A veces nos interesará tomar un trozo del fichero para hacer pruebas, por ejemplo las primeras 1000 secuencias (o 4000 líneas):
> zcat reads.fq.gz | head -4000 > test_reads.fq
> zcat reads.fq.gz | head -4000 | gzip > test_reads.fq.gz
O extraer un rango de líneas (1000001-1000004):
> zcat reads.fq.gz | sed -n '1000001,1000004p;1000005q' > lines.fq
Para extraer 1000 secuencias aleatorias de un archivo FASTQ:
> cat reads.fq | awk '{ printf("%s",$0); n++; if(n%4==0) { printf("\n");} else { printf("X#&X");} }' | shuf | head -1000 | sed 's/X#&X/\n/g' > reads.1000.fq
O de un archivo FASTA:
> cat reads.fa | awk '{if ((NR%2)==0)print prev"X#&X"$0;prev=$0;}' | shuf | head -1000 | sed 's/X#&X/\n/g' > reads.1000.fa
También nos puede interesar dividir el fichero en varios más pequeños de por ejemplo 1 miĺlón de secuencias (o 4 millones de líneas):
> zcat reads.fq.gz | split -d -l 4000000 - reads.split
> gzip reads.split*
> rename 's/\.split(\d+)/.$1.fq/' reads.split*
Y posteriormente reunificarlos:
> zcat reads.*.fq.gz | gzip > reads.fq.gz
Algunos de estos comandos y otras ideas pueden consultarse en:
http://darrenjw.wordpress.com/2010/11/28/introduction-to-the-processing-of-short-read-next-generation-sequencing-data/
Podéis ayudarme a completar el post escribiendo vuestros comandos más útiles en comentarios...
19 de diciembre de 2014
kseq::klib to parse FASTQ/FASTA files
Buenas,
éste tiene pinta de ser el último artículo del año, no? Desde hace tiempo venimos usando en el laboratorio la librería kseq.h , integrante de [ https://github.com/attractivechaos/klib ], para las desagradecidas tareas de manipular grandes archivos en formato FASTQ. Esta librería es un único archivo .h en lenguaje C que de manera muy eficiente permite leer archivos FASTQ, y también FASTA, incluso comprimidos con GZIP, con zlib. La eficiencia es fundamental para estas aplicaciones, porque modificar archivos .fastq de varios GB puede consumir mucho tiempo.
Como resultado de esta experiencia hace unos meses colgamos de la web del labo un programita, que llamamos split_pairs. Este software facilita trabajos tediosos como pueden ser modificar las cabeceras de una archivo FASTQ con expresiones regulares para que un programa posterior no se queje, y de paso emparejar o interlazar los reads según convenga. Está escrito en C++ y tiene asociado un script Perl.
De lo que quería hablar hoy es de que es posible utilizar kseq.h directamente desde nuestro lenguaje preferido. Para ellos es necesario emplear herramientas que permitan incluir, y compilar, código C mezclado con el de nuestro lenguaje interpretado. En el caso de Python estaríamos hablando por ejemplo de cython , aquí os muestro cómo hacerlo en Perl con el módulo Inline::CPP, dotando a kseq de una interfaz orientada a objetos para evitar pasar tipos de datos con tipos no estándar:
Ahora estamos en disposición de probar a leer un archivo con este módulo:
Qué ganancia en tiempo de procesamiento de archivos FASTQ se obtiene? Una prueba realizada con el módulo Benchmark , con diez repeticiones y un archivo .fastq.gz con 250K secuencias sugiere que kseq::klib es el doble de rápido:
s/iter perlio kseq
perlio 1.32 -- -50%
kseq 0.660 100% --
Hasta el próximo año, un saludo,
Bruno
éste tiene pinta de ser el último artículo del año, no? Desde hace tiempo venimos usando en el laboratorio la librería kseq.h , integrante de [ https://github.com/attractivechaos/klib ], para las desagradecidas tareas de manipular grandes archivos en formato FASTQ. Esta librería es un único archivo .h en lenguaje C que de manera muy eficiente permite leer archivos FASTQ, y también FASTA, incluso comprimidos con GZIP, con zlib. La eficiencia es fundamental para estas aplicaciones, porque modificar archivos .fastq de varios GB puede consumir mucho tiempo.
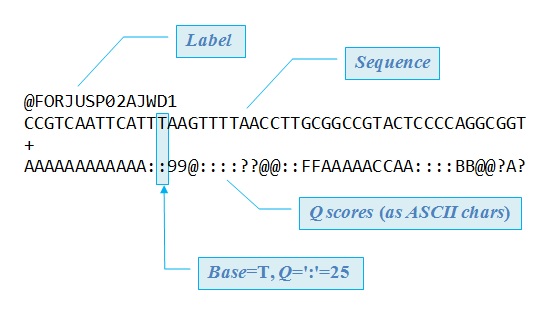 | |
| Fuente: http://drive5.com/usearch/manual/fastq_files.html |
Como resultado de esta experiencia hace unos meses colgamos de la web del labo un programita, que llamamos split_pairs. Este software facilita trabajos tediosos como pueden ser modificar las cabeceras de una archivo FASTQ con expresiones regulares para que un programa posterior no se queje, y de paso emparejar o interlazar los reads según convenga. Está escrito en C++ y tiene asociado un script Perl.
De lo que quería hablar hoy es de que es posible utilizar kseq.h directamente desde nuestro lenguaje preferido. Para ellos es necesario emplear herramientas que permitan incluir, y compilar, código C mezclado con el de nuestro lenguaje interpretado. En el caso de Python estaríamos hablando por ejemplo de cython , aquí os muestro cómo hacerlo en Perl con el módulo Inline::CPP, dotando a kseq de una interfaz orientada a objetos para evitar pasar tipos de datos con tipos no estándar:
package klib::kseq;
use Inline CPP => config => namespace => 'klib';
use Inline CPP => config => libs => '-lz';
use Inline CPP => <<'END_CPP';
/*
The Klib library [ https://github.com/attractivechaos/klib ], is a standalone and
lightweight C library distributed under MIT/X11 license.
Klib strives for efficiency and a small memory footprint.
This package contains C++ wrappers for components of Klib to make them easy to use from Perl scripts.
*/
using namespace std;
#include <iostream>
#include <string>
#include <zlib.h>
#include "kseq.h"
/*
kseq is generic stream buffer and a FASTA/FASTQ format parser from Klib.
Wrapper written by Bruno Contreras Moreira.
*/
KSEQ_INIT(gzFile, gzread)
class kseqreader
{
public:
kseqreader()
{
fp = NULL;
fromstdin=false;
openfile=false;
filename = "";
}
~kseqreader()
{
close_stream();
}
void rewind_stream()
{
if(openfile == true)
{
if(fromstdin == false) kseq_rewind(seq);
else
cerr<<"# reader: cannot rewind STDIN stream"<<endl;
}
}
void close_stream()
{
if(openfile == true)
{
kseq_destroy(seq);
if(fromstdin == false) gzclose(fp);
fp = NULL;
fromstdin=false;
openfile=false;
filename = "";
}
}
// accepts both FASTA and FASTQ files, which can be GZIP-compressed
// returns 1 if file was successfully opened, 0 otherwise
int open_stream(char *infilename)
{
filename = infilename;
if(filename.empty())
{
cerr<<"# reader: need a valid file name"<<endl;
filename = "";
return 0;
}
else
{
FILE *ifp = NULL;
if(filename == "stdin")
{
ifp = stdin;
fromstdin = true;
}
else ifp = fopen(filename.c_str(),"r");
if(ifp == NULL)
{
cerr<<"# reader: cannot open input file: "<<filename<<endl;
filename = "";
return 0;
}
else // open file and initialize stream
{
fp = gzdopen(fileno(ifp),"r");
seq = kseq_init(fp);
openfile=true;
return 1;
}
}
}
// returns length of current sequence, which can be 0,
// -1 in case of EOF and -2 in case of truncated quality string
int next_seq()
{
return kseq_read(seq);
}
const char *get_sequence()
{
return seq->seq.s;
}
// returns FASTA/FASTQ header up to first found space
const char *get_name()
{
return seq->name.s;
}
const char *get_quality()
{
if(seq->qual.l) return seq->qual.s;
else return "";
}
const char *get_comment()
{
if(seq->comment.l) return seq->comment.s;
else return "";
}
const char *get_full_header()
{
_header = seq->name.s;
if(seq->comment.l)
{
_header += " ";
_header + seq->comment.s;
}
return _header.c_str();
}
private:
gzFile fp;
kseq_t *seq;
bool fromstdin;
bool openfile;
string filename;
string _header;
};
END_CPP
1;
Ahora estamos en disposición de probar a leer un archivo con este módulo:
use strict;
use klib;
my ($length,$header,$sequence,$quality);
my $infile = "sample.fq.gz";
# my $infile = "stdin";
my $kseq = new klib::kseq::kseqreader();
if($kseq->open_stream($infile))
{
while( ($length = $kseq->next_seq()) != -1 )
{
($header,$sequence,$quality) =
($kseq->get_full_header(),$kseq->get_sequence(),$kseq->get_quality());
}
}
$kseq->close_stream()
Qué ganancia en tiempo de procesamiento de archivos FASTQ se obtiene? Una prueba realizada con el módulo Benchmark , con diez repeticiones y un archivo .fastq.gz con 250K secuencias sugiere que kseq::klib es el doble de rápido:
s/iter perlio kseq
perlio 1.32 -- -50%
kseq 0.660 100% --
Hasta el próximo año, un saludo,
Bruno
25 de noviembre de 2014
Una buena gráfica científica
Hola,
hoy quisiera compartir una imagen que demuestra, a pesar de toda la literatura al respecto, que no siempre es tan difícil hacer una buena gráfica con los datos de un experimento. En esta ocasión bastó con juntar todos los tomates cherry cosechados junto a la planta de la que proceden, y por supuesto una gráfica debajo para mostrar la varianza natural de estas plantas:
El artículo completo está en http://www.ncbi.nlm.nih.gov/pubmed/25362485 . Por cierto, tendrán algo de sabor esos tomates?
Hasta luego,
Bruno
hoy quisiera compartir una imagen que demuestra, a pesar de toda la literatura al respecto, que no siempre es tan difícil hacer una buena gráfica con los datos de un experimento. En esta ocasión bastó con juntar todos los tomates cherry cosechados junto a la planta de la que proceden, y por supuesto una gráfica debajo para mostrar la varianza natural de estas plantas:
 |
| Fuente: http://www.nature.com/ng/journal/v46/n12/fig_tab/ng.3131_F5.html. |
El artículo completo está en http://www.ncbi.nlm.nih.gov/pubmed/25362485 . Por cierto, tendrán algo de sabor esos tomates?
Hasta luego,
Bruno
Suscribirse a:
Entradas (Atom)