éste tiene pinta de ser el último artículo del año, no? Desde hace tiempo venimos usando en el laboratorio la librería kseq.h , integrante de [ https://github.com/attractivechaos/klib ], para las desagradecidas tareas de manipular grandes archivos en formato FASTQ. Esta librería es un único archivo .h en lenguaje C que de manera muy eficiente permite leer archivos FASTQ, y también FASTA, incluso comprimidos con GZIP, con zlib. La eficiencia es fundamental para estas aplicaciones, porque modificar archivos .fastq de varios GB puede consumir mucho tiempo.
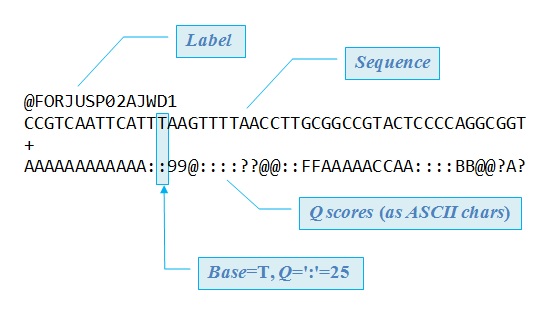 | |
| Fuente: http://drive5.com/usearch/manual/fastq_files.html |
Como resultado de esta experiencia hace unos meses colgamos de la web del labo un programita, que llamamos split_pairs. Este software facilita trabajos tediosos como pueden ser modificar las cabeceras de una archivo FASTQ con expresiones regulares para que un programa posterior no se queje, y de paso emparejar o interlazar los reads según convenga. Está escrito en C++ y tiene asociado un script Perl.
De lo que quería hablar hoy es de que es posible utilizar kseq.h directamente desde nuestro lenguaje preferido. Para ellos es necesario emplear herramientas que permitan incluir, y compilar, código C mezclado con el de nuestro lenguaje interpretado. En el caso de Python estaríamos hablando por ejemplo de cython , aquí os muestro cómo hacerlo en Perl con el módulo Inline::CPP, dotando a kseq de una interfaz orientada a objetos para evitar pasar tipos de datos con tipos no estándar:
package klib::kseq;
use Inline CPP => config => namespace => 'klib';
use Inline CPP => config => libs => '-lz';
use Inline CPP => <<'END_CPP';
/*
The Klib library [ https://github.com/attractivechaos/klib ], is a standalone and
lightweight C library distributed under MIT/X11 license.
Klib strives for efficiency and a small memory footprint.
This package contains C++ wrappers for components of Klib to make them easy to use from Perl scripts.
*/
using namespace std;
#include <iostream>
#include <string>
#include <zlib.h>
#include "kseq.h"
/*
kseq is generic stream buffer and a FASTA/FASTQ format parser from Klib.
Wrapper written by Bruno Contreras Moreira.
*/
KSEQ_INIT(gzFile, gzread)
class kseqreader
{
public:
kseqreader()
{
fp = NULL;
fromstdin=false;
openfile=false;
filename = "";
}
~kseqreader()
{
close_stream();
}
void rewind_stream()
{
if(openfile == true)
{
if(fromstdin == false) kseq_rewind(seq);
else
cerr<<"# reader: cannot rewind STDIN stream"<<endl;
}
}
void close_stream()
{
if(openfile == true)
{
kseq_destroy(seq);
if(fromstdin == false) gzclose(fp);
fp = NULL;
fromstdin=false;
openfile=false;
filename = "";
}
}
// accepts both FASTA and FASTQ files, which can be GZIP-compressed
// returns 1 if file was successfully opened, 0 otherwise
int open_stream(char *infilename)
{
filename = infilename;
if(filename.empty())
{
cerr<<"# reader: need a valid file name"<<endl;
filename = "";
return 0;
}
else
{
FILE *ifp = NULL;
if(filename == "stdin")
{
ifp = stdin;
fromstdin = true;
}
else ifp = fopen(filename.c_str(),"r");
if(ifp == NULL)
{
cerr<<"# reader: cannot open input file: "<<filename<<endl;
filename = "";
return 0;
}
else // open file and initialize stream
{
fp = gzdopen(fileno(ifp),"r");
seq = kseq_init(fp);
openfile=true;
return 1;
}
}
}
// returns length of current sequence, which can be 0,
// -1 in case of EOF and -2 in case of truncated quality string
int next_seq()
{
return kseq_read(seq);
}
const char *get_sequence()
{
return seq->seq.s;
}
// returns FASTA/FASTQ header up to first found space
const char *get_name()
{
return seq->name.s;
}
const char *get_quality()
{
if(seq->qual.l) return seq->qual.s;
else return "";
}
const char *get_comment()
{
if(seq->comment.l) return seq->comment.s;
else return "";
}
const char *get_full_header()
{
_header = seq->name.s;
if(seq->comment.l)
{
_header += " ";
_header + seq->comment.s;
}
return _header.c_str();
}
private:
gzFile fp;
kseq_t *seq;
bool fromstdin;
bool openfile;
string filename;
string _header;
};
END_CPP
1;
Ahora estamos en disposición de probar a leer un archivo con este módulo:
use strict;
use klib;
my ($length,$header,$sequence,$quality);
my $infile = "sample.fq.gz";
# my $infile = "stdin";
my $kseq = new klib::kseq::kseqreader();
if($kseq->open_stream($infile))
{
while( ($length = $kseq->next_seq()) != -1 )
{
($header,$sequence,$quality) =
($kseq->get_full_header(),$kseq->get_sequence(),$kseq->get_quality());
}
}
$kseq->close_stream()
Qué ganancia en tiempo de procesamiento de archivos FASTQ se obtiene? Una prueba realizada con el módulo Benchmark , con diez repeticiones y un archivo .fastq.gz con 250K secuencias sugiere que kseq::klib es el doble de rápido:
s/iter perlio kseq
perlio 1.32 -- -50%
kseq 0.660 100% --
Hasta el próximo año, un saludo,
Bruno
















{kind=link}