La idea básica es que los alumnos puedan usar python para sus trabajos
diarios y dejen de usar herramientas con licencias pagas (matlab,origin, etc).
Éste es el temario, y los ejercicios están en el link al final de la pág:
https://github.com/pewen/cpc
Abrazo,
Daniel
23 de septiembre de 2014
apuntes de python científico
Me envía mi colega Daniel Bellomo de la UNRC, en Argentina, un enlace a una recopilación de material didáctico que están usando los alumnos de Químicas para aprender a ser autónomos en cálculo científico con software Open Source, iniciativa que apoyamos y por eso difundimos también aquí:
9 de septiembre de 2014
contrato en mejora y genómica de cebada
El Departamento de Genética y Producción Vegetal de la Estación Experimental de Aula Dei – CSIC ofrece un contrato de 4 años para la realización de una tesis doctoral ligada al proyecto AGL2013-48756-R “Descubrimiento y utilización de la variabilidad genética que determina la adaptación de la cebada mediante herramientas genéticas y genómicas”.
El trabajo persigue el descubrimiento de caracteres y genes que ayuden a la obtención de variedades de cebada mejoradas para condiciones de sequía. Consiste en una combinación de los más modernos enfoques genéticos (diversidad intraespecífica, mapeo de QTL en poblaciones de cebada), genómicos (marcadores por métodos de secuenciación) y bioinformáticos (integración de datos de captura de exoma y otras fuentes de secuencia). El resultado de este trabajo permitirá identificar regiones cromosómicas y, potencialmente, genes de variedades tradicionales de cebada que contribuirán directamente a la mejora de variedades (el grupo también participa en un programa de mejora de variedades). Además, estos estudios permitirán profundizar en la naturaleza de los procesos de adaptación de las plantas al clima, contribuyendo en general a la mejora de variedades para condiciones de cambio climático.
Los candidatos deben tener vocación por la investigación, un buen expediente académico, excelente nivel de inglés, y deben estar admitidos en un programa de doctorado al finalizar el plazo de subsanación de la convocatoria. Se valorará tener cursado un master oficial relacionado con el tema de trabajo y/o experiencia en bioinformática.
Las solicitudes deberán ser presentadas por los candidatos del 10 de septiembre de al 26 de septiembre de 2014 a las 15:00 horas (hora peninsular española). Se ruega a las personas interesadas se pongan en contacto cuanto antes con los investigadores responsables (acasas at eead.csic.es, igartua at eead.csic.es, pgracia at eead.csic.es, bcontreras at eead.csic.es) para enviar una carta de interés y una copia del CV.
Referencias relevantes:
http://www.eead.csic.es/EEAD/barley
http://floresta.eead.csic.es/barleymap
El trabajo persigue el descubrimiento de caracteres y genes que ayuden a la obtención de variedades de cebada mejoradas para condiciones de sequía. Consiste en una combinación de los más modernos enfoques genéticos (diversidad intraespecífica, mapeo de QTL en poblaciones de cebada), genómicos (marcadores por métodos de secuenciación) y bioinformáticos (integración de datos de captura de exoma y otras fuentes de secuencia). El resultado de este trabajo permitirá identificar regiones cromosómicas y, potencialmente, genes de variedades tradicionales de cebada que contribuirán directamente a la mejora de variedades (el grupo también participa en un programa de mejora de variedades). Además, estos estudios permitirán profundizar en la naturaleza de los procesos de adaptación de las plantas al clima, contribuyendo en general a la mejora de variedades para condiciones de cambio climático.
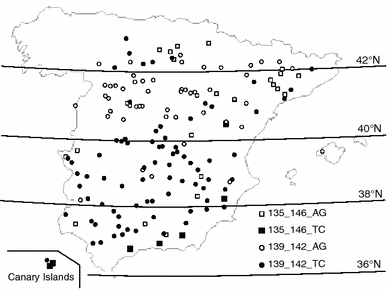 | |
| Distribución de haplotipos de HvFT1 en líneas de la Colección Nuclear de Cebadas Españolas colectas en la Península Ibérica. Tomada de http://link.springer.com/article/10.1007%2Fs00122-011-1531-x. |
Los candidatos deben tener vocación por la investigación, un buen expediente académico, excelente nivel de inglés, y deben estar admitidos en un programa de doctorado al finalizar el plazo de subsanación de la convocatoria. Se valorará tener cursado un master oficial relacionado con el tema de trabajo y/o experiencia en bioinformática.
Las solicitudes deberán ser presentadas por los candidatos del 10 de septiembre de al 26 de septiembre de 2014 a las 15:00 horas (hora peninsular española). Se ruega a las personas interesadas se pongan en contacto cuanto antes con los investigadores responsables (acasas at eead.csic.es, igartua at eead.csic.es, pgracia at eead.csic.es, bcontreras at eead.csic.es) para enviar una carta de interés y una copia del CV.
Referencias relevantes:
http://www.eead.csic.es/EEAD/barley
http://floresta.eead.csic.es/barleymap
11 de julio de 2014
blast eficiente con alias de nr
Hola,
en esta entrada quisiera mostraros una manera de buscar secuencias de proteínas dentro de la base de datos estrictamente no redundante nr del NCBI, restringiendo la búsqueda a grupos taxonómicos arbitrarios, sin necesidad de duplicar secuencias en otros archivos. De hecho, por su gran tamaño, 45 millones de secuencias a dia de hoy, desde hace tiempo nosotros ya no trabajamos con el archivo FASTA de nr, si no con la base de datos ya formateada para blast que descargamos regularmente con update_blastdb.pl desde ftp://ftp.ncbi.nlm.nih.gov/blast/db .
La receta para realizar búsquedas sobre subconjuntos de nr la hemos sacado de https://www.biostars.org/p/6528, y aquí os mostraré un ejemplo con secuencias de plantas:
1) Si no la tienes ya descárgate con el script update_blastdb.pl una copia de nr. Ojo, son una serie de archivos que en total ocupan 42G:
$ perl update_blastdb.pl nr
2) Encontrar el identificador taxonómico que mejor represente al subconjunto de nr que quieres definir, en nuestro caso Viridiplantae:
http://www.ncbi.nlm.nih.gov/Taxonomy/Browser/wwwtax.cgi?id=33090

3) Buscar todas las proteínas de plantas anotadas en el NCBI y exportar (Send to File => GI List) sus identificadores de GenBank (GIs) a un archivo, por ejemplo 'Viridiplantae_protein.gi.txt':
http://www.ncbi.nlm.nih.gov/protein/?term=txid33090[ORGN]

4) Hacer un alias de nr que contenga sólo proteínas de plantas:
$ ncbi-blast-2.2.29+/bin/blastdb_aliastool -gilist Viridiplantae_protein.gi.txt -db nr -out nr_plants -title nr_plants
En mi ejemplo produce el siguiente output:
Converted 3741334 GIs from Viridiplantae_protein.gi.txt to binary format in nr_plants.p.gil
Created protein BLAST (alias) database nr_plants with 2041649 sequences
Ahora veamos qué diferencias y ventajas tiene usar este subconjunto de nr en comparación con la base de datos completa:
[ NR ]
$ time ncbi-blast-2.2.29+/bin/blastx -query test.fna -db nr
[ NR_plants ]
$ time ncbi-blast-2.2.29+/bin/blastx -query test.fna -db nr_plants
Además, el tiempo de búsqueda con un sólo procesador es del orden de 20x más rápido con el alias nr_plants, y a la vez el consumo de RAM en tiempo real mucho menor.
Hasta luego,
Bruno
en esta entrada quisiera mostraros una manera de buscar secuencias de proteínas dentro de la base de datos estrictamente no redundante nr del NCBI, restringiendo la búsqueda a grupos taxonómicos arbitrarios, sin necesidad de duplicar secuencias en otros archivos. De hecho, por su gran tamaño, 45 millones de secuencias a dia de hoy, desde hace tiempo nosotros ya no trabajamos con el archivo FASTA de nr, si no con la base de datos ya formateada para blast que descargamos regularmente con update_blastdb.pl desde ftp://ftp.ncbi.nlm.nih.gov/blast/db .
La receta para realizar búsquedas sobre subconjuntos de nr la hemos sacado de https://www.biostars.org/p/6528, y aquí os mostraré un ejemplo con secuencias de plantas:
1) Si no la tienes ya descárgate con el script update_blastdb.pl una copia de nr. Ojo, son una serie de archivos que en total ocupan 42G:
$ perl update_blastdb.pl nr
2) Encontrar el identificador taxonómico que mejor represente al subconjunto de nr que quieres definir, en nuestro caso Viridiplantae:
http://www.ncbi.nlm.nih.gov/Taxonomy/Browser/wwwtax.cgi?id=33090
3) Buscar todas las proteínas de plantas anotadas en el NCBI y exportar (Send to File => GI List) sus identificadores de GenBank (GIs) a un archivo, por ejemplo 'Viridiplantae_protein.gi.txt':
http://www.ncbi.nlm.nih.gov/protein/?term=txid33090[ORGN]
4) Hacer un alias de nr que contenga sólo proteínas de plantas:
$ ncbi-blast-2.2.29+/bin/blastdb_aliastool -gilist Viridiplantae_protein.gi.txt -db nr -out nr_plants -title nr_plants
En mi ejemplo produce el siguiente output:
Converted 3741334 GIs from Viridiplantae_protein.gi.txt to binary format in nr_plants.p.gil
Created protein BLAST (alias) database nr_plants with 2041649 sequences
Ahora veamos qué diferencias y ventajas tiene usar este subconjunto de nr en comparación con la base de datos completa:
[ NR ]
$ time ncbi-blast-2.2.29+/bin/blastx -query test.fna -db nr
Database: All non-redundant GenBank CDS translations+PDB+SwissProt+PIR+PRF excluding environmental samples from WGS projects 45,360,603 sequences; 16,206,973,370 total letters Query= test Length=312 Score E Sequences producing significant alignments: (Bits) Value dbj|BAK07057.1| predicted protein [Hordeum vulgare subsp. vulgare] 156 8e-47 dbj|BAJ89957.1| predicted protein [Hordeum vulgare subsp. vulgar... 62.0 5e-10 ref|WP_014745933.1| asparagine synthase [Tistrella mobilis] >ref 42.7 0.020 ref|WP_006720063.1| hypothetical protein [Collinsella stercoris]... 35.0 5.0 ref|XP_001817163.2| hypothetical protein AOR_1_2924174 [Aspergil... 35.0 5.3 dbj|BAE55161.1| unnamed protein product [Aspergillus oryzae RIB40] 35.0 6.9 ref|WP_021178774.1| Phosphate transport system permease protein ... 34.7 7.6 ref|WP_021807128.1| Phosphate transport system permease protein ... 34.7 7.9 ref|WP_026166948.1| glucan biosynthesis protein D [Novosphingobi... 34.3 8.8 [...] real 5m28.923s user 5m24.564s sys 0m3.596s
[ NR_plants ]
$ time ncbi-blast-2.2.29+/bin/blastx -query test.fna -db nr_plants
Database: nr_plants 2,041,649 sequences; 772,729,335 total letters Query= test Length=312 Score E Sequences producing significant alignments: (Bits) Value dbj|BAK07057.1| predicted protein [Hordeum vulgare subsp. vulgare] 156 4e-48 dbj|BAJ89957.1| predicted protein [Hordeum vulgare subsp. vulgar... 62.0 2e-11 gb|EMS57556.1| hypothetical protein TRIUR3_28581 [Triticum urartu] 33.1 0.95 ref|XP_002311179.2| hypothetical protein POPTR_0008s05850g [Popu... 33.5 1.1 ref|XP_004509944.1| PREDICTED: uncharacterized protein LOC101513... 32.0 3.0 ref|XP_004509945.1| PREDICTED: uncharacterized protein LOC101513... 32.0 3.0 gb|EMT15944.1| hypothetical protein F775_17477 [Aegilops tauschii] 32.0 3.3 dbj|BAK00277.1| predicted protein [Hordeum vulgare subsp. vulgare] 32.0 3.7 ref|XP_004234300.1| PREDICTED: uncharacterized protein LOC101244... 31.2 4.7 ref|XP_006358717.1| PREDICTED: LRR receptor-like serine/threonin... 31.2 5.2 ref|XP_002316273.2| hypothetical protein POPTR_0010s20870g [Popu... 31.2 6.6 ref|XP_003571199.1| PREDICTED: probable tocopherol cyclase, chlo... 30.8 8.4 ref|XP_008451998.1| PREDICTED: respiratory burst oxidase homolog... 30.4 9.9 [...] real 0m16.805s user 0m16.332s sys 0m0.372sComo se puede ver en ambos casos las dos primeras secuencias encontradas son las mismas, con la misma puntuación en bits y distinto E-valor, al ser universos de búsqueda de tamaños distintos.
Además, el tiempo de búsqueda con un sólo procesador es del orden de 20x más rápido con el alias nr_plants, y a la vez el consumo de RAM en tiempo real mucho menor.
Hasta luego,
Bruno
9 de julio de 2014
regenerando DNA degenerado
Hola,
durante la reciente visita de mi colega Pablo Vinuesa al laboratorio pasamos ratos escribiendo código en Perl con el fin de diseñar de manera automática, para grandes conjuntos de secuencias de DNA previamente alineadas, parejas de primers (cebadores de PCR) adecuadas para estudios de microbiología ambiental, siguiendo principios bien conocidos ya. En cuanto probamos el código con unos cuantos ejemplos observamos que a menudo los primers para algunas regiones de los alineamientos múltiples eran degenerados. Como se muestra en la figura, eso significa que algunas posiciones de los cebadores no podían ser nucleótidos únicos si no que tenían que ser combinaciones de 2 o más para poder hibridarse con las secuencias utlizadas para su diseño.
A la hora de evaluar parejas de primers nos encontramos con que los degenerados en realidad tendrían que ser examinados por medio de cada una de las secuencias implícitas en el código degenerado. Y por tanto, tuvimos que buscar una manera de regenerar las secuencias de nucleótidos correspondientes a un primer degenerado, que es de lo que va esta entrada. El siguiente código en Perl hace precisamente esto:
Podemos probarlo con las secuencias de la figura:
$ perl degenprimer.pl GAYTST
GACTCT
GATTCT
GACTGT
GATTGT
y
$ perl degenprimer.pl GHGKAG
GAGGAG
GCGGAG
GTGGAG
GAGTAG
GCGTAG
GTGTAG
Hasta otra,
Bruno
durante la reciente visita de mi colega Pablo Vinuesa al laboratorio pasamos ratos escribiendo código en Perl con el fin de diseñar de manera automática, para grandes conjuntos de secuencias de DNA previamente alineadas, parejas de primers (cebadores de PCR) adecuadas para estudios de microbiología ambiental, siguiendo principios bien conocidos ya. En cuanto probamos el código con unos cuantos ejemplos observamos que a menudo los primers para algunas regiones de los alineamientos múltiples eran degenerados. Como se muestra en la figura, eso significa que algunas posiciones de los cebadores no podían ser nucleótidos únicos si no que tenían que ser combinaciones de 2 o más para poder hibridarse con las secuencias utlizadas para su diseño.
 | |
| Pareja de primers degenerados que definen un amplicón a partir de secuencias de DNA alineadas. De acuerdo con la nomenclatura IUPAC, el de la izquierda (fwd) puede escribirse como GAYTST y el de la derecha (rev) como GHGKAG. Figura tomada de http://acgt.cs.tau.ac.il/hyden. |
#!/usr/bin/perl
use strict;
my $degenerate_sequence = $ARGV[0] || die "# usage: $0 <degenerate DNA sequence>\n";
my $regenerated_seqs = regenerate($degenerate_sequence);
foreach my $seq (@$regenerated_seqs)
{
print "$seq\n";
}
# returns a ref to list of all DNA sequences implied in a degenerate oligo
# adapted from Amplicon (Simon Neil Jarman, http://sourceforge.net/projects/amplicon)
sub regenerate
{
my ($primerseq) = @_;
my %IUPACdegen = (
'A'=>['A'],'C'=>['C'], 'G'=>['G'], 'T'=>['T'],
'R'=>['A','G'], 'Y'=>['C','T'], 'S'=>['C','G'], 'W'=>['A','T'], 'K'=>['G','T'], 'M'=>['A','C'],
'B'=>['C','G','T'], 'D'=>['A','G','T'], 'V'=>['A','C','G'], 'H'=>['A','C','T'],
'N'=>['A','C','G','T']
);
my @oligo = split(//,uc($primerseq));
my @grow = ('');
my @newgrow = ('');
my ($res,$degen,$x,$n,$seq);
foreach $res (0 .. $#oligo){
$degen = $IUPACdegen{$oligo[$res]};
if($#{$degen}>0){
$x = 0;
@newgrow = @grow;
while($x<$#{$degen}){
push(@newgrow,@grow);
$x++;
}
@grow = @newgrow;
}
$n=$x=0;
foreach $seq (0 .. $#grow){
$grow[$seq] .= $degen->[$x];
$n++;
if($n == (scalar(@grow)/scalar(@$degen))){
$n=0;
$x++;
}
}
}
return \@grow;
}
Podemos probarlo con las secuencias de la figura:
$ perl degenprimer.pl GAYTST
GACTCT
GATTCT
GACTGT
GATTGT
y
$ perl degenprimer.pl GHGKAG
GAGGAG
GCGGAG
GTGGAG
GAGTAG
GCGTAG
GTGTAG
Hasta otra,
Bruno
17 de junio de 2014
postdoc computational systems biology Luxemburg
Postdoctoral Fellow in Computational Systems Biology at the Luxembourg Centre for Systems Biomedicine in collaboration with the Black Family Stem Cell Institute of the Mount Sinai School of Medicine
http://www.nature.com/naturejobs/science/jobs/425291-postdoctoral-fellow-in-systems-biologyThe Computational Biology Group seeks a highly skilled and motivated Postdotoral Fellow to work on an exciting project on the application of network biology approaches to study the role of gene expression stochasticity in cell fate commitment. In particular, the selected candidate shall develop a computational model that integrates transcriptomics and epigenomics data to describe heterogeneity in embryonic stem cells and its implication in differentiation. Single cell and cell perturbation experiments will be performed in order to validate the predictions generated by the model. This project will be carried out in collaboration with Profs. Ihor Lemischka and Kateri Moore at the Black Family Stem Cell Institute of the Mount Sinai School of Medicine. The selected applicant will have the opportunity to visit the experimental labs of our collaborators at the Mount Sinai School of Medicine.
Requirements of the ideal candidate:
- Ph.D. in Bioinformatics, Computer Science, Biology or a related discipline
- Strong computational skills
- Prior experience in mathematical modelling of biological networks, especially in network inference and analysis
- A strong first-author publication record in the fields of Bioinformatics and Computational Biology
- Excellent working knowledge in English.
We offer:
- Opportunity to do applied research to medical problems within a highly dynamic research institution (LCSB) and in collaboration with internationally recognized partners
- An exciting international environment
- A very competitive salary
For further information, please contact:
Prof. Dr. Antonio del Sol, Luxembourg Centre for Systems BiomedicineE-mail: antonio.delsol@uni.lu
Suscribirse a:
Entradas (Atom)