Hi,
late last year we published a paper describing GET_PANGENES, a protocol to call pangenes, which are clusters of gene models/alleles found in genomic assemblies in a similar location. You can read all about it at https://doi.org/10.1186/s13059-023-03071-z . Using this approach you can produce figures like this, where you can see the pangene of interest in green:
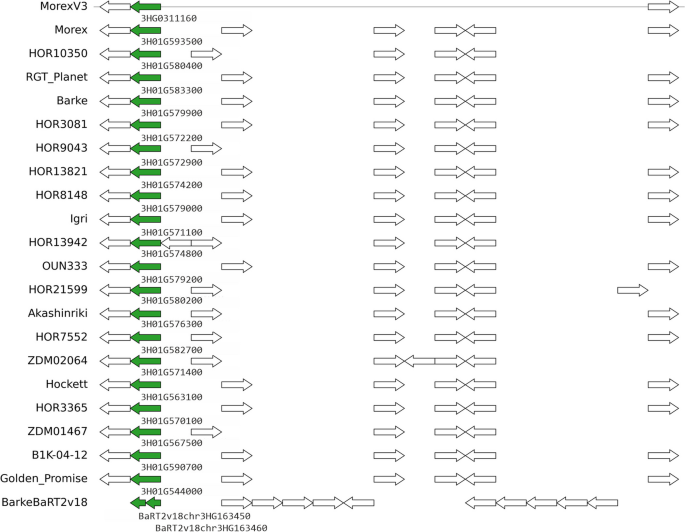 |
| Genomic context of barly pangene cluster HORVU.MOREX.r3.3HG0311160 (green arrows), which corresponds to barley locus HvOS2. Figure from https://doi.org/10.1186/s13059-023-03071-z |
As we do research on barley breeding and adaptation, we thought it would be useful for us and others out there to have way of inspecting barley pangenes, for instance to check whether a gene of interest is conserved or polymorphic across the barleys sampled by in the pangenome (n=20) put together by Jayakodi et al.
This exactly what you can do, at the protein sequence level, at https://eead-csic-compbio.github.io/barley_pangenes , where you can scroll pangenes along chromosomes, with MorexV3 positions; genes not found in MorexV3 lack a position therefore and are shown with a hash (#):

You will notice that pangenes with occupancy > 1, ie containing gene models found it at least two barleys, can be clicked to display a multiple protein alignment with help from the NCBI msaviewer:

There you can easily zoom in to regions of interest and print or export the alignment in FASTA, PDF or SVG format (high quality).
Hope this can be useful to the barley genomics community,
Bruno