Secuenciación de amplicones (SA) es una traducción aproximada al español de la técnica de
"Amplicon sequencing" que junto con las tecnologías de secuenciación masiva (del inglés
new generation sequencing, NGS) permite genotipar cientos/miles de individuos en un único experimento.
La secuenciación de amplicones (SA) consiste en secuenciar los productos de múltiples PCRs. Un
amplicón se define como el conjunto de secuencias obtenidas de cada PCR individual.
Antiguamente se realizaban PCRs individuales y se secuenciaban uno a uno los productos. Con las nuevas técnicas de NGS, podemos incluir
etiquetas de DNA (por ej. una secuencia única de 6 nucleótidos)
diferentes para cientos de muestras o individuos y clasificar más tarde las secuencias o
reads resultantes de una única secuenciación (
Binladen et al. 2007;
Meyer et al. 2007).
 |
| Esquema de etiquetado y amplificación para la secuenciación de amplicones. |
Mediante esta técnica podremos
genotipar individuos y distinguir los diferentes alelos (con la secuenciación tradicional a veces es complicado separar alelos de un mismo gen). El principal problema de las técnicas de NGS es su alta tasa de error, que a su vez puede ser compensada incrementando la profundidad de secuenciación (el número de
reads). Otros problemas pueden ser los errores de la polimerasa o la generación de quimeras (una secuencia mezcla de otras).
En el siguiente enlace podemos ver un
vídeo explicativo del proceso de secuenciación de amplicones:
http://www.jove.com/video/51709/la-secuenciacin-de-prxima-generacin-de-16s-arn-ribosomal-genes?language=Spanish
Básicamente existen 4 etapas en el análisis por AS con NGS:
- Diseño experimental de los primers usados para amplificar los genes de interés (marcadores) y las etiquetas a usar para distinguir los diferentes individuos o muestras.
- Amplificación por PCR de los marcadores en el laboratorio, generalmente se realiza una PCR por cada muestra.
- Secuenciación de los productos de amplificación. Las tecnologías de NGS más usadas para SA son: Illumina, 454 e Ion Torrent.
- Análisis bioinformático de los datos de secuenciación. El análisis incluye separación de las reads en amplicones, corrección de errores de secuenciación, filtrado de reads minoritarias/contaminantes y generación de genotipados.
 |
| Etapas de la técnica de secuenciación de amplicones mediante NGS. |
Aplicaciones
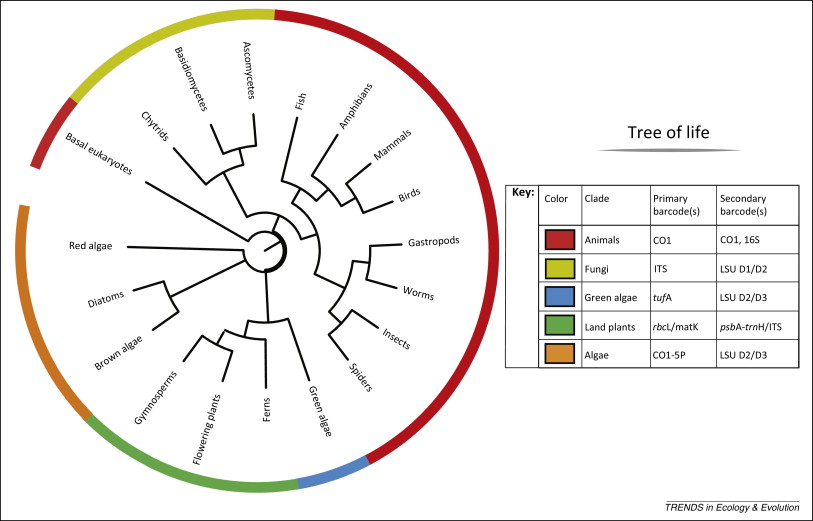
SA es utilizado para realizar
clasificaciones taxonómicas usando genes como:
cytochrome c oxidase subunit
1 (CO1), genes rRNA (16S/18S/28S), genes específicos de plantas (rbcL,
matK, and trnH-psbA) y espaciadores internos nucleares (ITSs)
(
Kress et al. 2014;
Joly et al. 2014).
Los genes anteriores se distinguen por una tasa de mutación
suficientemente rápida como para distinguir especies cercanas y a la vez
suficientemente estables como para distinguir congéneres.
 |
| Lista de genes habitualmente usados como marcadores taxonómicos (fuente: Kress et al. 2014) |
Un experimento pionero de SA fue la
determinación de la diversidad microbiana en aguas marinas profundas (
Sogin et al. 2006), usando primers flanquenado la región V6 hipervariable de la subunidad 16S rRNA bacteriana. Dicho estudio descubrió miles de poblaciones minoritarias de organismos no conocidos con anterioridad.
Otro gran campo de aplicación es el
genotipado de familias de genes de alta complejidad, como el complejo mayor de histocompatibilidad, que poseen múltiples
loci y diferente número de copias entre individuos, incluso de la misma especie (
Babik et al. 2010;
Lighten et al. 2014). El
complejo mayor de histocompatibilidad (MHC) de clase I y II codifica receptores celulares que presentan antígenos a las células del sistema inmune y son los genes más polimórficos conocidos en vertebrados . El MHC humano también se conoce como
HLA (Human Leukocyte Antigen) y juega un papel clave en la compatibilidad en el transplante de órganos. Los
loci del MHC son tan polimórficos que no hay dos individuos en una población no endogámica que posean el mismo conjunto de alelos (excepto gemelos).
Hasta hace poco era necesario
clonar y secuenciar uno por uno los diferentes alelos de este tipo de genes para conseguir una secuencia fiable. Actualmente tan tediosa tarea puede ser simplificada mediante
un único experimento de NGS que incluya múltiples individuos y múliples genes. El secuenciador de nueva generación (ej.: Illumina, 454 o Ion Torrent) leerá las sequencias individuales de cada uno de los alelos. Actualmente existen incluso kits comerciales para simplificar el proceso: Illumina TruSeq Custom
Amplicon, Roche 454 Fluidigm Access Array or Life Technologies Ion
Torrent Ion AmpliSeq.